Charlotte Dravet | |
|---|---|
| Born | 14 July 1936 |
Charlotte Dravet (born July 14, 1936) is a French paediatric psychiatrist and epileptologist.
Charlotte Dravet | |
|---|---|
| Born | 14 July 1936 |
Charlotte Dravet (born July 14, 1936) is a French paediatric psychiatrist and epileptologist.
After graduation at the Aix-Marseille University, Dravet trained in Pediatrics in Marseille, France from 1962–1965. She wrote her M.D. thesis on the Lennox-Gastaut syndrome. [1] In 1971 she was certified as psychiatrist.
From 1965 to 2000, Dravet specialized in epilepsy at the Centre Saint Paul in Marseille, among others with Henri Gastaut, Joseph Roger, and René Soulayrol (pediatric psychiatry). [2] She was the resident doctor and actually lived on the premises until her retirement in 2000. Dravet had the opportunity, accompany and observe inpatients for many years, which resulted in some of her major contributions to epileptology.
In 1972, Dravet trained in the pediatric EEG Department of the Hôpital Saint-Vincent de Paul and in the Department of Functional Neurosurgery of the Hôpital Sainte-Anne in Paris. From 1989 to 2000, Dravet was Associate Medical Director of the Centre Saint Paul.
With Joseph Roger and Michelle Bureau, Dravet played an active role in the delineation of epileptic syndromes through several workshops and the first edition of the book "Epileptic syndromes in infancy, childhood and adolescence". In 1981 she described together with Michelle Bureau the benign myoclonic epilepsy of infancy [3] and in 1978 [4] as well as in 1982 the severe myoclonic epilepsy of childhood. This syndrome was later named the Dravet syndrome, [5] [6] which was confirmed by subsequent genetic discoveries and became a model for the genetic childhood epilepsies.
From 1991 to 1993, Charlotte Dravet was a member of the Scientific Board of the French Foundation for Research on Epilepsy. From 1996 to 2004, she was a member of the Task Force on Classification and Terminology of the International League Against Epilepsy ILAE. From 1997 to 1999, she served as president of the French League Against Epilepsy (LFCE). In 2000 she organized the first National Epilepsy Day in France. From 2003 to 2006, she was a member of the Board of the French Comité National pour l’Épilepsie and she still is a member of the Scientific Board of the International Dravet syndrome Epilepsy Action League.
Since her retirement, Dravet has focused her activities on the Dravet syndrome. As Honorary Consultant she regularly attends the Childhood Epilepsy Unit at the Policlinico A. Gemelli of the Università Cattolica del Sacro Cuore in Rome, Italy, where she sees patients with this severe epilepsy and, in collaboration with her Italian colleagues, coordinates research on their cognitive development.
Dravet trained or helped train many epileptologists who came to Marseille to learn about the epileptic syndromes of infancy and childhood. She is also a frequent speaker about epilepsy and has spoken at epilepsy meetings and workshops worldwide. Dravet has also participate in events organised by patients’ and parents’ associations worldwide.
Charlotte Dravet is Honorary Member of several chapters of the ILAE. She has been awarded as "Ambassador for Epilepsy" by the ILAE and the International Bureau for Epilepsy (IBE) in 1989, with the European Epileptology Prize by the Commission on European Affairs (CEA) of the ILAE in 2004, and with the "Lifetime Achievement Award" by the ILAE and IBE in 2017. [8] In 2019, she received the Dravet Award [9] from the Dravet Syndrome Foundation Spain for her contribution to the knowledge and research of Dravet syndrome. In 2011 she has been nominated Chevalier in the French Order of the Légion d'honneur.

Epilepsy is a group of non-communicable neurological disorders characterized by recurrent epileptic seizures. An epileptic seizure is the clinical manifestation of an abnormal, excessive, and synchronized electrical discharge in the brain cells called neurons. The occurrence of two or more unprovoked seizures defines epilepsy. The occurrence of just one seizure may warrant the definition in a more clinical usage where recurrence may be able to be prejudged. Epileptic seizures can vary from brief and nearly undetectable periods to long periods of vigorous shaking due to abnormal electrical activity in the brain. These episodes can result in physical injuries, either directly such as broken bones or through causing accidents. In epilepsy, seizures tend to recur and may have no detectable underlying cause. Isolated seizures that are provoked by a specific cause such as poisoning are not deemed to represent epilepsy. People with epilepsy may be treated differently in various areas of the world and experience varying degrees of social stigma due to the alarming nature of their symptoms.
Absence seizures are one of several kinds of generalized seizures. In the past, absence epilepsy was referred to as "pyknolepsy," a term derived from the Greek word "pyknos," signifying "extremely frequent" or "grouped". These seizures are sometimes referred to as petit mal seizures ; however, usage of this terminology is no longer recommended. Absence seizures are characterized by a brief loss and return of consciousness, generally not followed by a period of lethargy. Absence seizures are most common in children. They affect both sides of the brain.
Henri Jean Pascal Gastaut was a French neurologist and epileptologist.
Lennox–Gastaut syndrome (LGS) is a complex, rare, and severe childhood-onset epilepsy syndrome. It is characterized by multiple and concurrent seizure types including tonic seizure, cognitive dysfunction, and slow spike waves on electroencephalogram (EEG), which are very abnormal. Typically, it presents in children aged 3–5 years and most of the time persists into adulthood with slight changes in the electroclinical phenotype. It has been associated with perinatal injuries, congenital infections, brain malformations, brain tumors, genetic disorders such as tuberous sclerosis and numerous gene mutations. Sometimes LGS is observed after infantile epileptic spasm syndrome. The prognosis for LGS is marked by a 5% mortality in childhood and persistent seizures into adulthood.
Non-epileptic seizures (NES), also known as non-epileptic events, are paroxysmal events that appear similar to an epileptic seizure but do not involve abnormal, rhythmic discharges of neurons in the brain. Symptoms may include shaking, loss of consciousness, and loss of bladder control.
William Gordon Lennox was an American neurologist and epileptologist who was a pioneer in the use of electroencephalography (EEG) for the diagnosis and treatment of epilepsy. He graduated from Colorado College and Harvard Medical School.
In the field of neurology, seizure types are categories of seizures defined by seizure behavior, symptoms, and diagnostic tests. The International League Against Epilepsy (ILAE) 2017 classification of seizures is the internationally recognized standard for identifying seizure types. The ILAE 2017 classification of seizures is a revision of the prior ILAE 1981 classification of seizures. Distinguishing between seizure types is important since different types of seizures may have different causes, outcomes, and treatments.

Frederick Andermann was a Canadian neurologist and epileptologist.
Dravet syndrome (DS), previously known as severe myoclonic epilepsy of infancy (SMEI), is an autosomal dominant genetic disorder which causes a catastrophic form of epilepsy, with prolonged seizures that are often triggered by hot temperatures or fever. It is very difficult to treat with anticonvulsant medications. It often begins before one year of age, with six months being the age that seizures, characterized by prolonged convulsions and triggered by fever, usually begin.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).

Chromodomain-helicase-DNA-binding protein 2 is an enzyme that in humans is encoded by the CHD2 gene.
Jean Aicardi was a French pediatric neurologist and epileptologist. He was known as one of the most distinguished and respected neuropediatricians of his time. He, along with Alexis Arzimanoglou, created the journal Epileptic Disorders in 1999.
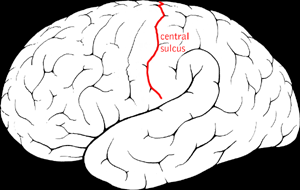
Benign Rolandic epilepsy or self-limited epilepsy with centrotemporal spikes is the most common epilepsy syndrome in childhood. Most children will outgrow the syndrome, hence the label benign. The seizures, sometimes referred to as sylvian seizures, start around the central sulcus of the brain.
Panayiotopoulos syndrome is a common idiopathic childhood-related seizure disorder that occurs exclusively in otherwise normal children and manifests mainly with autonomic epileptic seizures and autonomic status epilepticus. An expert consensus has defined Panayiotopoulos syndrome as "a benign age-related focal seizure disorder occurring in early and mid-childhood. It is characterized by seizures, often prolonged, with predominantly autonomic symptoms, and by an EEG [electroencephalogram] that shows shifting and/or multiple foci, often with occipital predominance."
Migralepsy is a rare condition in which a migraine is followed, within an hour period, by an epileptic seizure. Because of the similarities in signs, symptoms, and treatments of both conditions, such as the neurological basis, the psychological issues, and the autonomic distress that is created from them, they individually increase the likelihood of causing the other. However, also because of the sameness, they are often misdiagnosed for each other, as migralepsy rarely occurs.

Infantile convulsions and choreoathetosis (ICCA) syndrome is a neurological genetic disorder with an autosomal dominant mode of inheritance. It is characterized by the association of benign familial infantile epilepsy (BIFE) at age 3–12 months and later in life with paroxysmal kinesigenic choreoathetosis. The ICCA syndrome was first reported in 1997 in four French families from north-western France and provided the first genetic evidence for common mechanisms shared by benign infantile seizures and paroxysmal dyskinesia. The epileptic origin of PKC has long been a matter of debates and PD have been classified as reflex epilepsies. Indeed, attacks of PKC and epileptic seizures have several characteristics in common, they both are paroxysmal in presentation with a tendency to spontaneous remission, and a subset of PKC responds well to anticonvulsants. This genetic disease has been mapped to chromosome 16p-q12. More than 30 families with the clinical characteristics of ICCA syndrome have been described worldwide so far.
Epilepsy-intellectual disability in females also known as PCDH19 gene-related epilepsy or epileptic encephalopathy, early infantile, 9 (EIEE9), is a rare type of epilepsy that affects predominately females and is characterized by clusters of brief seizures, which start in infancy or early childhood, and is occasionally accompanied by varying degrees of cognitive impairment. The striking pattern of onset seizures at a young age, genetic testing and laboratory results, potential developmental delays or developmental regression and associated disorders, eases diagnosis.
People with epilepsy may be classified into different syndromes based on specific clinical features. These features include the age at which seizures begin, the seizure types, and EEG findings, among others. Identifying an epilepsy syndrome is useful as it helps determine the underlying causes as well as deciding what anti-seizure medication should be tried. Epilepsy syndromes are more commonly diagnosed in infants and children. Some examples of epilepsy syndromes include benign rolandic epilepsy, childhood absence epilepsy and juvenile myoclonic epilepsy. Severe syndromes with diffuse brain dysfunction caused, at least partly, by some aspect of epilepsy, are also referred to as epileptic encephalopathies. These are associated with frequent seizures that are resistant to treatment and severe cognitive dysfunction, for instance Lennox-Gastaut syndrome and West syndrome.

Ingrid Eileen Scheffer is an Australian paediatric neurologist and senior research fellow at the Florey Institute of Neuroscience and Mental Health. She has made several major advances in the field of epilepsy research. Scheffer is credited with finding the first gene implicated in epilepsy. She has also described and classified novel epileptic syndromes such as Epilepsy limited to Females with Mental Retardation.
Malignant migrating partial seizures of infancy (MMPSI) is a rare epileptic syndrome that onsets before 6 months of age, commonly in the first few weeks of life. Once seizures start, the site of seizure activity repeatedly migrates from one area of the brain to another, with few periods of remission in between. These seizures are 'focal' (updated term for 'partial'), meaning they do not affect both sides of the brain at the same time. These continuous seizures cause damage to the brain, hence the descriptor 'malignant.'
| International | |
|---|---|
| National | |
| Other | |