
Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data, in particular when the data sets are large and complex. As an interdisciplinary field of science, bioinformatics combines biology, chemistry, physics, computer science, information engineering, mathematics and statistics to analyze and interpret the biological data. Bioinformatics has been used for in silico analyses of biological queries using computational and statistical techniques.

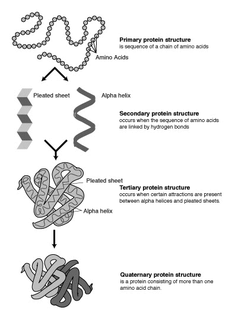
Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

Protein folding is the physical process by which a protein chain is translated to its native three-dimensional structure, typically a "folded" conformation by which the protein becomes biologically functional. Via an expeditious and reproducible process, a polypeptide folds into its characteristic three-dimensional structure from a random coil. Each protein exists first as an unfolded polypeptide or random coil after being translated from a sequence of mRNA to a linear chain of amino acids. At this stage the polypeptide lacks any stable (long-lasting) three-dimensional structure. As the polypeptide chain is being synthesized by a ribosome, the linear chain begins to fold into its three-dimensional structure.

In biochemistry, allosteric regulation is the regulation of an enzyme by binding an effector molecule at a site other than the enzyme's active site.

The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland, and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.

In molecular biology, molecular chaperones are proteins that assist the conformational folding or unfolding of large proteins or macromolecular protein complexes. There are a number of classes of molecular chaperones, all of which function to assist large proteins in proper protein folding during or after synthesis, and after partial denaturation. Chaperones are also involved in the translocation of proteins for proteolysis.

Membrane proteins are common proteins that are part of, or interact with, biological membranes. Membrane proteins fall into several broad categories depending on their location. Integral membrane proteins are a permanent part of a cell membrane and can either penetrate the membrane (transmembrane) or associate with one or the other side of a membrane. Peripheral membrane proteins are transiently associated with the cell membrane.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanics force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, the monomers of the polymer. A single amino acid monomer may also be called a residue indicating a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo electron microscopy (cryo-EM) and dual polarisation interferometry to determine the structure of proteins.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.

In molecular biology, an intrinsically disordered protein (IDP) is a protein that lacks a fixed or ordered three-dimensional structure, typically in the absence of its macromolecular interaction partners, such as other proteins or RNA. IDPs range from fully unstructured to partially structured and include random coil, molten globule-like aggregates, or flexible linkers in large multi-domain proteins. They are sometimes considered as a separate class of proteins along with globular, fibrous and membrane proteins.
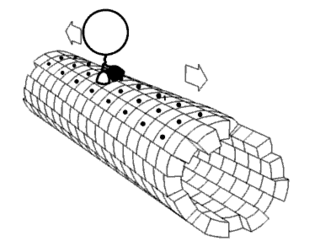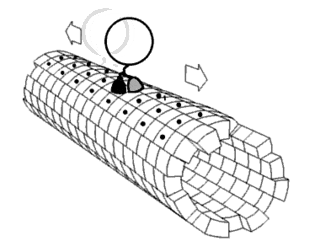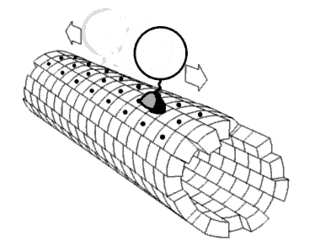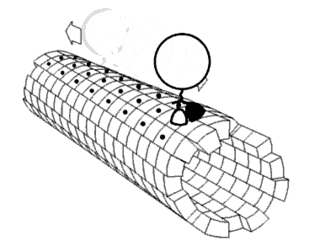
In biochemistry, a conformational change is a change in the shape of a macromolecule, often induced by environmental factors.
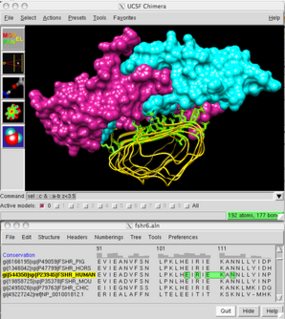
UCSF Chimera is an extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. High-quality images and movies can be created. Chimera includes complete documentation and can be downloaded free of charge for noncommercial use.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.

In molecular biology, a protein domain is a region of a protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains, and a domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length. The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.
In molecular modelling, docking is a method which predicts the preferred orientation of one molecule to another when bound together in a stable complex. In the case of protein docking, the search space consists of all possible orientations of the protein with respect to the ligand. Flexible docking in addition considers all possible conformations of the protein paired with all possible conformations of the ligand.
Proteins are generally thought to adopt unique structures determined by their amino acid sequences. However, proteins are not strictly static objects, but rather populate ensembles of conformations. Transitions between these states occur on a variety of length scales and time scales, and have been linked to functionally relevant phenomena such as allosteric signaling and enzyme catalysis.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.