
Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.
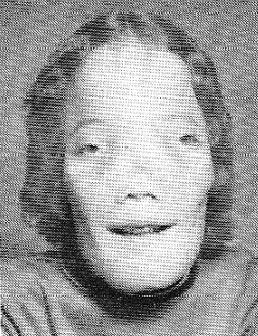
Roy Lee "Rocky" Dennis was an American teenager who had craniodiaphyseal dysplasia, an extremely rare sclerotic bone disorder. The condition usually results in neurological disorders and death during childhood or teenage years. His life was the basis for the 1985 drama film Mask.

Spondyloperipheral dysplasia is an autosomal dominant disorder of bone growth. The condition is characterized by flattened bones of the spine (platyspondyly) and unusually short fingers and toes (brachydactyly). Some affected individuals also have other skeletal abnormalities, short stature, nearsightedness (myopia), hearing loss, and mental retardation. Spondyloperipheral dysplasia is a subtype of collagenopathy, types II and XI.
Spondyloepiphyseal dysplasia congenita is a rare disorder of bone growth that results in dwarfism, characteristic skeletal abnormalities, and occasionally problems with vision and hearing. The name of the condition indicates that it affects the bones of the spine (spondylo-) and the ends of bones (epiphyses), and that it is present from birth (congenital). The signs and symptoms of spondyloepiphyseal dysplasia congenita are similar to, but milder than, the related skeletal disorders achondrogenesis type 2 and hypochondrogenesis. Spondyloepiphyseal dysplasia congenita is a subtype of collagenopathy, types II and XI.

Autosomal recessive multiple epiphyseal dysplasia (ARMED), also called epiphyseal dysplasia, multiple, 4 (EDM4), multiple epiphyseal dysplasia with clubfoot or –with bilayered patellae, is an autosomal recessive congenital disorder affecting cartilage and bone development. The disorder has relatively mild signs and symptoms, including joint pain, scoliosis, and malformations of the hands, feet, and knees.
Papillorenal syndrome is an autosomal dominant genetic disorder marked by underdevelopment (hypoplasia) of the kidney and colobomas of the optic nerve.
Dermatopathia pigmentosa reticularis(DPR) is a rare, autosomal dominant congenital disorder that is a form of ectodermal dysplasia. Dermatopathia pigmentosa reticularis is composed of the triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy. DPR is a non life-threatening disease that largely affects the skin, hair, and nails. It has also been identified as a keratin disorder. Historically, as of 1992, only 10 cases had been described in world literature; however, due to recent advances in genetic analysis, five additional families studied in 2006 have been added to the short list of confirmed cases.
Rosselli–Gulienetti syndrome, also known as Zlotogora–Ogur syndrome and Bowen–Armstrong syndrome, is a type of congenital ectodermal dysplasia syndrome. The syndrome is relatively rare and has only been described in a few cases.

Spondylocostal dysostosis, also known as Jarcho-Levin syndrome (JLS), is a rare, heritable axial skeleton growth disorder. It is characterized by widespread and sometimes severe malformations of the vertebral column and ribs, shortened thorax, and moderate to severe scoliosis and kyphosis. Individuals with Jarcho-Levin typically appear to have a short trunk and neck, with arms appearing relatively long in comparison, and a slightly protuberant abdomen. Severely affected individuals may have life-threatening pulmonary complications due to deformities of the thorax. The syndrome was first described by Saul Jarcho and Paul M. Levin at Johns Hopkins University in 1938.
Fibrochondrogenesis is a rare autosomal recessive form of osteochondrodysplasia, causing abnormal fibrous development of cartilage and related tissues.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
Potocki–Shaffer syndrome (PSS), also known as DEFECT11 syndrome or chromosome 11p11.2 deletion syndrome, is a rare contiguous gene syndrome that results from the microdeletion of section 11.2 on the short arm of chromosome 11 (11p11.2). The syndrome has its name from Dr. Lorraine (Lori) Potocki and Dr. Lisa Shaffer who discovered the deletion on the 11th chromosome and studied the impacts.
Opsismodysplasia is a type of skeletal dysplasia first described by Zonana and associates in 1977, and designated under its current name by Maroteaux (1984). Derived from the Greek opsismos ("late"), the name "opsismodysplasia" describes a delay in bone maturation. In addition to this delay, the disorder is characterized by micromelia, particularly of the hands and feet, delay of ossification, platyspondyly, irregular metaphyses, an array of facial aberrations and respiratory distress related to chronic infection. Opsismodysplasia is congenital, being apparent at birth. It has a variable mortality, with some affected individuals living to adulthood. The disorder is rare, with an incidence of less than 1 per 1,000,000 worldwide. It is inherited in an autosomal recessive pattern, which means the defective (mutated) gene that causes the disorder is located on an autosome, and the disorder occurs when two copies of this defective gene are inherited. No specific gene has been found to be associated with the disorder. It is similar to spondylometaphyseal dysplasia, Sedaghatian type.
Parastremmatic dwarfism is a rare bone disease that features severe dwarfism, thoracic kyphosis, a distortion and twisting of the limbs, contractures of the large joints, malformations of the vertebrae and pelvis, and incontinence. The disease was first reported in 1970 by Leonard Langer and associates; they used the term parastremmatic from the Greek parastremma, or distorted limbs, to describe it. On X-rays, the disease is distinguished by a "flocky" or lace-like appearance to the bones. The disease is congenital, which means it is apparent at birth. It is caused by a mutation in the TRPV4 gene, located on chromosome 12 in humans. The disease is inherited in an autosomal dominant manner.
Kohlschütter–Tönz syndrome (KTS), also called amelo-cerebro-hypohidrotic syndrome, is a rare inherited syndrome characterized by epilepsy, psychomotor delay or regression, intellectual disability, and yellow teeth caused by amelogenesis imperfecta. It is a type A ectodermal dysplasia.
Kenny-Caffey syndrome type 2 (KCS2) is an extremely rare autosomal dominant genetic condition characterized by dwarfism, hypermetropia, microphthalmia, and skeletal abnormalities. This subtype of Kenny-Caffey syndrome is caused by a heterozygous mutation in the FAM111A gene (615292) on chromosome 11q12.
Czech dysplasia metatarsal type is a rare type of Czech dysplasia which is characterized primarily by bone anomalies.
Schneckenbecken dysplasia is a rare pre-natally fatal hereditary autosomal recessive condition which affects the bones and pre-natal growth.

Calvarial doughnut lesions-bone fragility syndrome, also known as familial calvarial doughnut lesions, is a rare autosomal dominant genetic disorder characterized by mild to moderate fragility of the bones accompanied with calvarial doughnut lesions.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
