
QDPR is a human gene that produces the enzyme quinoid dihydropteridine reductase. This enzyme is part of the pathway that recycles a substance called tetrahydrobiopterin, also known as BH4. Tetrahydrobiopterin works with an enzyme called phenylalanine hydroxylase to process a substance called phenylalanine. Phenylalanine is an amino acid that is obtained through the diet; it is found in all proteins and in some artificial sweeteners. When tetrahydrobiopterin interacts with phenylalanine hydroxylase, tetrahydrobiopterin is altered and must be recycled to a usable form. The regeneration of this substance is critical for the proper processing of several other amino acids in the body. Tetrahydrobiopterin also helps produce certain chemicals in the brain called neurotransmitters, which transmit signals between nerve cells.

Cytochrome P450 1B1 is an enzyme that in humans is encoded by the CYP1B1 gene.

Lecithin–cholesterol acyltransferase is an enzyme, in many animals including humans, that converts free cholesterol into cholesteryl ester, which is then sequestered into the core of a lipoprotein particle, eventually making the newly synthesized HDL spherical and forcing the reaction to become unidirectional since the particles are removed from the surface. The enzyme is bound to high-density lipoproteins (HDLs) (alpha-LCAT) and LDLs (beta-LCAT) in the blood plasma. LCAT deficiency can cause impaired vision due to cholesterol corneal opacities, anemia, and kidney damage. It belongs to the family of phospholipid:diacylglycerol acyltransferases.

7-Dehydrocholesterol reductase, also known as DHCR7, is a protein that in humans is encoded by the DHCR7 gene.

Familial hypercholesterolemia (FH) is a genetic disorder characterized by high cholesterol levels, specifically very high levels of low-density lipoprotein, in the blood and early cardiovascular disease.The most common mutations diminish the number of functional LDL receptors in the liver. Since the underlying body biochemistry is slightly different in individuals with FH, their high cholesterol levels are less responsive to the kinds of cholesterol control methods which are usually more effective in people without FH. Nevertheless, treatment is usually effective.
N-acetylgalactosamine-6-sulfatase is an enzyme that, in humans, is encoded by the GALNS gene.
The human gene SRD5A2 encodes the 3-oxo-5α-steroid 4-dehydrogenase 2 enzyme, also known as 5α-reductase type 2 (5αR2), one of three isozymes of 5α-reductase.

Sepiapterin reductase is an enzyme that in humans is encoded by the SPR gene.

The CLCN family of voltage-dependent chloride channel genes comprises nine members which demonstrate quite diverse functional characteristics while sharing significant sequence homology. The protein encoded by this gene regulates the electric excitability of the skeletal muscle membrane. Mutations in this gene cause two forms of inherited human muscle disorders: recessive generalized myotonia congenita (Becker) and dominant myotonia (Thomsen).

Bifunctional UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase is an enzyme that in humans is encoded by the GNE gene.

Exostosin-1 is a protein that in humans is encoded by the EXT1 gene.

Phosphorylase b kinase regulatory subunit alpha, liver isoform is an enzyme that in humans is encoded by the PHKA2 gene.

Sialin, also known as H(+)/nitrate cotransporter and H(+)/sialic acid cotransporter, is a protein which in humans is encoded by the SLC17A5 gene.
Sterol-4-alpha-carboxylate 3-dehydrogenase, decarboxylating is an enzyme that in humans is encoded by the NSDHL gene. This enzyme is localized in the endoplasmic reticulum and is involved in cholesterol biosynthesis.
Protein hairless is a protein that in humans is encoded by the HR gene.

24-Dehydrocholesterol reductase is a protein that in humans is encoded by the DHCR24 gene.
Chitobiosyldiphosphodolichol beta-mannosyltransferase is an enzyme that is encoded by ALG1 whose structure and function has been conserved from lower to higher organisms.

Lathosterolosis is a defect in cholesterol biosynthesis.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.
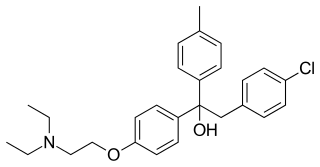
Triparanol was the first synthetic cholesterol-lowering drug. It was patented in 1959 and introduced in the United States in 1960. The developmental code name of triparanol, MER/29, became so well known that it became the registered trade name of the drug. It was withdrawn in 1962 due to severe adverse effects such as nausea and vomiting, vision loss due to irreversible cataracts, alopecia, skin disorders, and accelerated atherosclerosis. It is now considered to be obsolete.
