
Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder affecting about one in 2,500 people.

Spinal muscular atrophies (SMAs) are a genetically and clinically heterogeneous group of rare debilitating disorders characterised by the degeneration of lower motor neurons and subsequent atrophy (wasting) of various muscle groups in the body. While some SMAs lead to early infant death, other diseases of this group permit normal adult life with only mild weakness.

Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a progressive debilitating neurodegenerative disorder resulting in muscle cramps and progressive weakness due to degeneration of motor neurons in the brainstem and spinal cord.
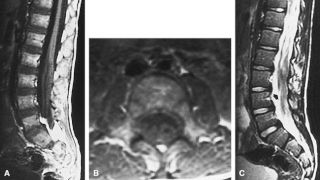
Dejerine–Sottas disease, also known as, Dejerine–Sottas neuropathy, progressive hypertrophic interstitial polyneuropathy of childhood and onion bulb neuropathy, is a hereditary neurological disorder characterised by damage to the peripheral nerves and resulting progressive muscle wasting. The condition is caused by mutations in a various genes and currently has no known cure.

Myelin protein zero is a single membrane glycoprotein which in humans is encoded by the MPZ gene. P0 is a major structural component of the myelin sheath in the peripheral nervous system (PNS). Myelin protein zero is expressed by Schwann cells and accounts for over 50% of all proteins in the peripheral nervous system, making it the most common protein expressed in the PNS. Mutations in myelin protein zero can cause myelin deficiency and are associated with neuropathies like Charcot–Marie–Tooth disease and Dejerine–Sottas disease.

Heat shock protein beta-8 is a protein that in humans is encoded by the HSPB8 gene.

Glycine—tRNA ligase also known as glycyl–tRNA synthetase is an enzyme that in humans is encoded by the GARS1 gene.

Surfeit locus protein 1 (SURF1) is a protein that in humans is encoded by the SURF1 gene. The protein encoded by SURF1 is a component of the mitochondrial translation regulation assembly intermediate of cytochrome c oxidase complex, which is involved in the regulation of cytochrome c oxidase assembly. Defects in this gene are a cause of Leigh syndrome, a severe neurological disorder that is commonly associated with systemic cytochrome c oxidase deficiency, and Charcot-Marie-Tooth disease 4K (CMT4K).

Hereditary motor and sensory neuropathies (HMSN) is a name sometimes given to a group of different neuropathies which are all characterized by their impact upon both afferent and efferent neural communication. HMSN are characterised by atypical neural development and degradation of neural tissue. The two common forms of HMSN are either hypertrophic demyelinated nerves or complete atrophy of neural tissue. Hypertrophic condition causes neural stiffness and a demyelination of nerves in the peripheral nervous system, and atrophy causes the breakdown of axons and neural cell bodies. In these disorders, a patient experiences progressive muscle atrophy and sensory neuropathy of the extremities.
Hereditary sensory and autonomic neuropathy (HSAN) or hereditary sensory neuropathy (HSN) is a condition used to describe any of the types of this disease which inhibit sensation.

Distal spinal muscular atrophy type 1 (DSMA1), also known as spinal muscular atrophy with respiratory distress type 1 (SMARD1), is a rare neuromuscular disorder involving death of motor neurons in the spinal cord which leads to a generalised progressive atrophy of body muscles.

Hereditary neuropathy with liability to pressure palsy (HNPP) is a peripheral neuropathy, a condition that affects the nerves. Pressure on the nerves can cause tingling sensations, numbness, pain, weakness, muscle atrophy and even paralysis of the affected area. In normal individuals, these symptoms disappear quickly, but in sufferers of HNPP even a short period of pressure can cause the symptoms to occur. Palsies can last from minutes or days to weeks or even months.
Distal hereditary motor neuronopathies, sometimes also called distal hereditary motor neuropathies, are a genetically and clinically heterogeneous group of motor neuron diseases that result from genetic mutations in various genes and are characterized by degeneration and loss of motor neuron cells in the anterior horn of the spinal cord and subsequent muscle atrophy.

Spinal muscular atrophy with lower extremity predominance 1 (SMALED1) is an extremely rare neuromuscular disorder of infants characterised by severe progressive muscle atrophy which is especially prominent in legs.
Roussy–Lévy syndrome, also known as Roussy–Lévy hereditary areflexic dystasia, is a rare genetic disorder of humans that results in progressive muscle wasting. It is caused by mutations in the genes that code for proteins necessary for the functioning of the myelin sheath of the neurons, affecting the conductance of nerve signals and resulting in loss of muscles' ability to move.
Hereditary motor and sensory neuropathy with proximal dominance (HMSN-P) is an autosomal dominant neurodegenerative disorder that is defined by extensive involuntary and spontaneous muscle contractions, asthenia, and atrophy with distal sensory involvement following. The disease starts presenting typically in the 40s and is succeeded by a slow and continuous onslaught. Muscle spasms and muscle contractions large in number are noted, especially in the earliest stages. The presentation of HMSN-P is quite similar to amyotrophic lateral sclerosis and has common neuropathological findings. Sensory loss happens as the disease progresses, but the amount of sensation lost varies from case to case. There have been other symptoms of HMSN-P reported such as urinary disturbances and a dry cough.
Distal spinal muscular atrophy type 2 (DSMA2), also known as Jerash type distal hereditary motor neuropathy (HMNJ), is a very rare childhood-onset genetic disorder characterised by progressive muscle wasting affecting lower and subsequently upper limbs. The disorder has been described in Arab inhabitants of Jerash region in Jordan as well as in a Chinese family.
Hereditary sensory and autonomic neuropathy type I or hereditary sensory neuropathy type I is a group of autosomal dominant inherited neurological diseases that affect the peripheral nervous system particularly on the sensory and autonomic functions. The hallmark of the disease is the marked loss of pain and temperature sensation in the distal parts of the lower limbs. The autonomic disturbances, if present, manifest as sweating abnormalities.
Seipin is a homo-oligomeric integral membrane protein in the endoplasmic reticulum (ER) that concentrates at junctions with cytoplasmic lipid droplets (LDs). Alternatively, seipin can be referred to as Bernardinelli-Seip congenital lipodystrophy type 2 protein (BSCL2), and it is encoded by the corresponding gene of the same name, i.e. BSCL2. At protein level, seipin is expressed in cortical neurons in the frontal lobes, as well as motor neurons in the spinal cord. It is highly expressed in areas like the brain, testis and adipose tissue. Seipin's function is still unclear but it has been localized close to lipid droplets, and cells knocked out in seipin which have anomalous droplets. Hence, recent evidence suggests that seipin plays a crucial role in lipid droplet biogenesis.
Mary M. Reilly FRCP is an Irish neurologist who works at National Hospital for Neurology and Neurosurgery. She studies peripheral neuropathy. She is the President of the Association of British Neurologists.
