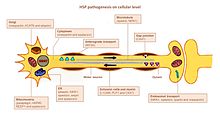
Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).

Pelizaeus–Merzbacher disease is an X-linked neurological disorder that damages oligodendrocytes in the central nervous system. It is caused by mutations in proteolipid protein 1 (PLP1), a major myelin protein. It is characterized by a decrease in the amount of insulating myelin surrounding the nerves (hypomyelination) and belongs to a group of genetic diseases referred to as leukodystrophies.
Spasticity is a feature of altered skeletal muscle performance with a combination of paralysis, increased tendon reflex activity, and hypertonia. It is also colloquially referred to as an unusual "tightness", stiffness, or "pull" of muscles.
Dystonia is a neurological hyperkinetic movement disorder in which sustained or repetitive muscle contractions occur involuntarily, resulting in twisting and repetitive movements or abnormal fixed postures. The movements may resemble a tremor. Dystonia is often intensified or exacerbated by physical activity, and symptoms may progress into adjacent muscles.
MASA syndrome is a rare X-linked recessive neurological disorder on the L1 disorder spectrum belonging in the group of hereditary spastic paraplegias a paraplegia known to increase stiffness spasticity in the lower limbs. This syndrome also has two other names, CRASH syndrome and Gareis-Mason syndrome.

L1, also known as L1CAM, is a transmembrane protein member of the L1 protein family, encoded by the L1CAM gene. This protein, of 200-220 kDa, is a neuronal cell adhesion molecule with a strong implication in cell migration, adhesion, neurite outgrowth, myelination and neuronal differentiation. It also plays a key role in treatment-resistant cancers due to its function. It was first identified in 1984 by M. Schachner who found the protein in post-mitotic mice neurons.

Machado–Joseph disease (MJD), also known as Machado–Joseph Azorean disease, Machado's disease, Joseph's disease or spinocerebellar ataxia type 3 (SCA3), is a rare autosomal dominantly inherited neurodegenerative disease that causes progressive cerebellar ataxia, which results in a lack of muscle control and coordination of the upper and lower extremities. The symptoms are caused by a genetic mutation that results in an expansion of abnormal "CAG" trinucleotide repeats in the ATXN3 gene that results in an abnormal form of the protein ataxin which causes degeneration of cells in the hindbrain. Some symptoms, such as clumsiness and rigidity, make MJD commonly mistaken for drunkenness or Parkinson's disease.
Hypertonia is a term sometimes used synonymously with spasticity and rigidity in the literature surrounding damage to the central nervous system, namely upper motor neuron lesions. Impaired ability of damaged motor neurons to regulate descending pathways gives rise to disordered spinal reflexes, increased excitability of muscle spindles, and decreased synaptic inhibition. These consequences result in abnormally increased muscle tone of symptomatic muscles. Some authors suggest that the current definition for spasticity, the velocity-dependent over-activity of the stretch reflex, is not sufficient as it fails to take into account patients exhibiting increased muscle tone in the absence of stretch reflex over-activity. They instead suggest that "reversible hypertonia" is more appropriate and represents a treatable condition that is responsive to various therapy modalities like drug or physical therapy.
Primary lateral sclerosis (PLS) is a very rare neuromuscular disease characterized by progressive muscle weakness in the voluntary muscles. PLS belongs to a group of disorders known as motor neuron diseases. Motor neuron diseases develop when the nerve cells that control voluntary muscle movement degenerate and die, causing weakness in the muscles they control.

The human gene SPAST codes for the microtubule-severing protein of the same name, commonly known as spastin.

Spartin is a protein that in humans is encoded by the SPG20 gene.
Leukoencephalopathy with neuroaxonal spheroids (LENAS), also known as adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD) is an extremely rare kind of leukoencephalopathy and is classified as a neurodegenerative disease. LENAS is a cause of severe and subacute dementia that results from damage to certain areas of the brain. This damage is to a type of brain tissue called white matter and axon damage due to swellings which are termed spheroids.

Kufor–Rakeb syndrome (KRS) is an autosomal recessive disorder of juvenile onset also known as Parkinson disease-9 (PARK9). It is named after Kufr Rakeb in Irbid, Jordan. Kufor–Rakeb syndrome was first identified in this region in Jordan with a Jordanian couple's 5 children who had rigidity, mask-like face, and bradykinesia. The disease was first described in 1994 by Najim Al-Din et al. The OMIM number is 606693.
Distal hereditary motor neuronopathies, sometimes also called distal hereditary motor neuropathies, are a genetically and clinically heterogeneous group of motor neuron diseases that result from genetic mutations in various genes and are characterized by degeneration and loss of motor neuron cells in the anterior horn of the spinal cord and subsequent muscle atrophy.
Hereditary sensory and autonomic neuropathy type I or hereditary sensory neuropathy type I is a group of autosomal dominant inherited neurological diseases that affect the peripheral nervous system particularly on the sensory and autonomic functions. The hallmark of the disease is the marked loss of pain and temperature sensation in the distal parts of the lower limbs. The autonomic disturbances, if present, manifest as sweating abnormalities.

L1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum dysgenesis, spastic paraplegia 1, MASA syndrome, and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS). It is also called L1CAM syndrome and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation, adducted thumbs, spasticity, and hydrocephalus.
Distal hereditary motor neuropathy type V is a particular type of neuropathic disorder. In general, distal hereditary motor neuropathies affect the axons of distal motor neurons and are characterized by progressive weakness and atrophy of muscles of the extremities. It is common for them to be called "spinal forms of Charcot-Marie-Tooth disease (CMT)", because the diseases are closely related in symptoms and genetic cause. The diagnostic difference in these diseases is the presence of sensory loss in the extremities. There are seven classifications of dHMNs, each defined by patterns of inheritance, age of onset, severity, and muscle groups involved. Type V is a disorder characterized by autosomal dominance, weakness of the upper limbs that is progressive and symmetrical, and atrophy of the small muscles of the hands.

Spinocerebellar ataxia type 1 (SCA1) is a rare autosomal dominant disorder, which, like other spinocerebellar ataxias, is characterized by neurological symptoms including dysarthria, hypermetric saccades, and ataxia of gait and stance. This cerebellar dysfunction is progressive and permanent. First onset of symptoms is normally between 30 and 40 years of age, though juvenile onset can occur. Death typically occurs within 10 to 30 years from onset.
Spastic paraplegia 15 (SPG15) is a form of hereditary spastic paraplegia that commonly becomes apparent during childhood or adolescence. The disease is caused by mutations within the ZFYVE26 gene - also known as the SPG15 gene - and is passed down in an autosomal recessive manner.
Spastic paraplegia 31 is a rare type of hereditary spastic paraplegia which is characterized by sensation anomalies of the lower extremities.