Barth syndrome (BTHS) is a rare but serious X-linked genetic disorder, caused by changes in phospholipid structure and metabolism. It may affect multiple body systems, and is potentially fatal. The syndrome is diagnosed almost exclusively in males.
Cardiolipin is an important component of the inner mitochondrial membrane, where it constitutes about 20% of the total lipid composition. It can also be found in the membranes of most bacteria. The name "cardiolipin" is derived from the fact that it was first found in animal hearts. It was first isolated from the beef heart in the early 1940s by Mary C. Pangborn. In mammalian cells, but also in plant cells, cardiolipin (CL) is found almost exclusively in the inner mitochondrial membrane, where it is essential for the optimal function of numerous enzymes that are involved in mitochondrial energy metabolism.

Tafazzin is a protein that in humans is encoded by the TAFAZZIN gene. Tafazzin is highly expressed in cardiac and skeletal muscle, and functions as a phospholipid-lysophospholipid transacylase. It catalyzes remodeling of immature cardiolipin to its mature composition containing a predominance of tetralinoleoyl moieties. Several different isoforms of the tafazzin protein are produced from the TAFAZZIN gene. A long form and a short form of each of these isoforms is produced; the short form lacks a hydrophobic leader sequence and may exist as a cytoplasmic protein rather than being membrane-bound. Other alternatively spliced transcripts have been described but the full-length nature of all these transcripts is not known. Most isoforms are found in all tissues, but some are found only in certain types of cells. Mutations in the TAFAZZIN gene have been associated with mitochondrial deficiency, Barth syndrome, dilated cardiomyopathy (DCM), hypertrophic DCM, endocardial fibroelastosis, left ventricular noncompaction (LVNC), breast cancer, papillary thyroid carcinoma, non-small cell lung cancer, glioma, gastric cancer, thyroid neoplasms, and rectal cancer.
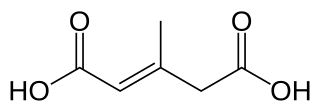
3-Methylglutaconic aciduria (MGA) is any of at least five metabolic disorders that impair the body's ability to make energy in the mitochondria. As a result of this impairment, 3-methylglutaconic acid and 3-methylglutaric acid build up and can be detected in the urine.
MT-ATP6 is a mitochondrial gene with the full name 'mitochondrially encoded ATP synthase membrane subunit 6' that encodes the ATP synthase Fo subunit 6. This subunit belongs to the Fo complex of the large, transmembrane F-type ATP synthase. This enzyme, which is also known as complex V, is responsible for the final step of oxidative phosphorylation in the electron transport chain. Specifically, one segment of ATP synthase allows positively charged ions, called protons, to flow across a specialized membrane inside mitochondria. Another segment of the enzyme uses the energy created by this proton flow to convert a molecule called adenosine diphosphate (ADP) to ATP. Mutations in the MT-ATP6 gene have been found in approximately 10 to 20 percent of people with Leigh syndrome.

85 kDa calcium-independent phospholipase A2, also known as 85/88 kDa calcium-independent phospholipase A2, Group VI phospholipase A2, Intracellular membrane-associated calcium-independent phospholipase A2 beta, or Patatin-like phospholipase domain-containing protein 9 is an enzyme that in humans is encoded by the PLA2G6 gene.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 8, mitochondrial also known as NADH-ubiquinone oxidoreductase 23 kDa subunit, Complex I-23kD (CI-23kD), or TYKY subunit is an enzyme that in humans is encoded by the NDUFS8 gene. The NDUFS8 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in this gene have been associated with Leigh syndrome.

Optic atrophy 3 protein is a protein that in humans is encoded by the OPA3 gene.
NADH dehydrogenase [ubiquinone] iron-sulfur protein 7, mitochondrial, also knowns as NADH-ubiquinone oxidoreductase 20 kDa subunit, Complex I-20kD (CI-20kD), or PSST subunit is an enzyme that in humans is encoded by the NDUFS7 gene. The NDUFS7 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain.
FAD-dependent oxidoreductase domain-containing protein 1 (FOXRED1), also known as H17, or FP634 is an enzyme that in humans is encoded by the FOXRED1 gene. FOXRED1 is an oxidoreductase and complex I-specific molecular chaperone involved in the assembly and stabilization of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in FOXRED1 have been associated with Leigh syndrome and infantile-onset mitochondrial encephalopathy.

Mitochondrial import inner membrane translocase subunit TIM14 is an enzyme that in humans is encoded by the DNAJC19 gene on chromosome 3. TIM14 belongs to the DnaJ family, which has been involved in Hsp40/Hsp70 chaperone systems. As a mitochondrial chaperone, TIM14 functions as part of the TIM23 complex import motor to facilitate the import of nuclear-encoded proteins into the mitochondria. TIM14 also complexes with prohibitin complexes to regulate mitochondrial morphogenesis, and has been implicated in dilated cardiomyopathy with ataxia.
Caseinolytic peptidase B protein homolog (CLPB), also known as Skd3, is a mitochondrial AAA ATPase chaperone that in humans is encoded by the gene CLPB, which encodes an adenosine triphosphate-(ATP) dependent chaperone. Skd3 is localized in mitochondria and widely expressed in human tissues. High expression in adult brain and low expression in granulocyte is found. It is a potent protein disaggregase that chaperones the mitochondrial intermembrane space. Mutations in the CLPB gene could cause autosomal recessive metabolic disorder with intellectual disability/developmental delay, congenital neutropenia, progressive brain atrophy, movement disorder, cataracts, and 3-methylglutaconic aciduria. Recently, heterozygous, dominant negative mutations in CLPB have been identified as a cause of severe congenital neutropenia (SCN).
Mitochondrial import inner membrane translocase subunit TIM50 is a protein that in humans is encoded by the TIMM50 gene. Tim50 is a subunit of the Tim23 translocase complex in the inner mitochondrial membrane. Mutations in TIMM50 can lead to epilepsy, severe intellectual disability, and 3-methylglutaconic aciduria. TIMM50 expression is increased in breast cancer cells and decreased in hypertrophic hearts.

Cytochrome c oxidase assembly protein COX15 homolog (COX15), also known as heme A synthase, is a protein that in humans is encoded by the COX15 gene. This protein localizes to the inner mitochondrial membrane and involved in heme A biosynthesis. COX15 is also part of a three-component mono-oxygenase that catalyses the hydroxylation of the methyl group at position eight of the protoheme molecule. Mutations in this gene has been reported in patients with hypertrophic cardiomyopathy as well as Leigh syndrome, and characterized by delayed onset of symptoms, hypotonia, feeding difficulties, failure to thrive, motor regression, and brain stem signs.
Solute carrier family 25 member 22 is a protein that in humans is encoded by the SLC25A22 gene. This gene encodes a mitochondrial glutamate carrier. Mutations in this gene are associated with early infantile epileptic encephalopathy. Expression of this gene is increased in colorectal tumor cells.
Chromosome 19 open reading frame 70, also known as QIL1, MICOS complex subunit MIC13 (MIC13) or Protein P117 is a protein that in humans is encoded by the C19orf70 gene.
Ubiquinol-cytochrome c reductase complex assembly factor 3 is a protein that in humans is encoded by the UQCC3 gene. Located in mitochondria, this protein is involved in the assembly of mitochondrial Complex III, stabilizing supercomplexes containing Complex III. Mutations in the UQCC3 gene cause Complex III deficiency with symptoms of hypoglycemia, hypotonia, lactic acidosis, and developmental delays. This protein plays an important role as an antiviral factor, bolstering the ability of cells to inhibit viral replication, independent of interferon production. The UQCC3 protein can be cleaved by OMA1 metalloprotease during mitochondrial depolarization, targeting the cell for apoptosis. Depletion of this protein alters cardiolipin composition, causing cellular and mitochondrial defects.
PET100 homolog is a protein that in humans is encoded by the PET100 gene. Mitochondrial complex IV, or cytochrome c oxidase, is a large transmembrane protein complex that is part of the respiratory electron transport chain of mitochondria. The small protein encoded by the PET100 gene plays a role in the biogenesis of mitochondrial complex IV. This protein localizes to the inner mitochondrial membrane and is exposed to the intermembrane space. Mutations in this gene are associated with mitochondrial complex IV deficiency. This gene has a pseudogene on chromosome 3. Alternative splicing results in multiple transcript variants.

Transmembrane protein 70 is a protein that in humans is encoded by the TMEM70 gene. It is a transmembrane protein located in the mitochondrial inner membrane involved in the assembly of the F1 and Fo structural subunits of ATP synthase. Mutations in this gene have been associated with neonatal mitochondrial encephalo-cardiomyopathy due to ATP synthase deficiency, causing a wide variety of symptoms including 3-methylglutaconic aciduria, lactic acidosis, mitochondrial myopathy, and cardiomyopathy.

Solute carrier family 25 member 46 is a protein that in humans is encoded by the SLC25A46 gene. This protein is a member of the SLC25 mitochondrial solute carrier family. It is a transmembrane protein located in the mitochondrial outer membrane involved in lipid transfer from the endoplasmic reticulum (ER) to mitochondria. Mutations in this gene result in neuropathy and optic atrophy.



