
Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.

Colpocephaly is a cephalic disorder involving the disproportionate enlargement of the occipital horns of the lateral ventricles and is usually diagnosed early after birth due to seizures. It is a nonspecific finding and is associated with multiple neurological syndromes, including agenesis of the corpus callosum, Chiari malformation, lissencephaly, and microcephaly. Although the exact cause of colpocephaly is not known yet, it is commonly believed to occur as a result of neuronal migration disorders during early brain development, intrauterine disturbances, perinatal injuries, and other central nervous system disorders. Individuals with colpocephaly have various degrees of motor disabilities, visual defects, spasticity, and moderate to severe intellectual disability. No specific treatment for colpocephaly exists, but patients may undergo certain treatments to improve their motor function or intellectual disability.

L1, also known as L1CAM, is a transmembrane protein member of the L1 protein family, encoded by the L1CAM gene. This protein, of 200-220 kDa, is a neuronal cell adhesion molecule with a strong implication in cell migration, adhesion, neurite outgrowth, myelination and neuronal differentiation. It also plays a key role in treatment-resistant cancers due to its function. It was first identified in 1984 by M. Schachner who found the protein in post-mitotic mice neurons.
Agenesis of the corpus callosum (ACC) is a rare birth defect in which there is a complete or partial absence of the corpus callosum. It occurs when the development of the corpus callosum, the band of white matter connecting the two hemispheres in the brain, in the embryo is disrupted. The result of this is that the fibers that would otherwise form the corpus callosum are instead longitudinally oriented along the ipsilateral ventricular wall and form structures called Probst bundles.
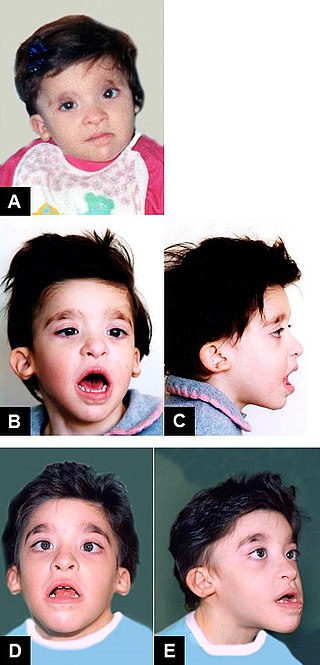
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998. The condition affects both males and females, has been described in various countries and ethnic groups around the world, and occurs in approximately 1 in 50,000-100,000 births.

SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. These characteristics are what make up the acronym SCARF. It shares some features with Lenz-Majewski hyperostotic dwarfism. It is a very rare disease with an incidence rate of approximately one in a million newborns. It has been clinically described in two males who were maternal cousins, as well as a 3-month-old female. Babies affected by this syndrome tend to have very loose skin, giving them an elderly facial appearance. Possible complications include dyspnea, abdominal hernia, heart disorders, joint disorders, and dislocations of multiple joints. It is believed that this disease's inheritance is X-linked recessive.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.
Vici syndrome, also called immunodeficiency with cleft lip/palate, cataract, hypopigmentation and absent corpus callosum, is a rare autosomal recessive congenital disorder characterized by albinism, agenesis of the corpus callosum, cataracts, cardiomyopathy, severe psychomotor retardation, seizures, immunodeficiency and recurrent severe infections. To date, about 50 cases have been reported.

Acrocallosal syndrome is an extremely rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms. The syndrome was first described by Albert Schinzel in 1979. Mutations in KIF7 are causative for ACLS, and mutations in GLI3 are associated with a similar syndrome.
FG syndrome (FGS) is a rare genetic syndrome caused by one or more recessive genes located on the X chromosome and causing physical anomalies and developmental delays. FG syndrome was named after the first letters of the surnames of the first patients noted with the disease. First reported by American geneticists John M. Opitz and Elisabeth G. Kaveggia in 1974, its major clinical features include intellectual disability, hyperactivity, hypotonia, and a characteristic facial appearance including macrocephaly.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
Smith–Fineman–Myers syndrome (SFMS1) is a congenital disorder that causes birth defects. This syndrome was named after Richard D. Smith, Robert M. Fineman and Gart G. Myers who discovered it around 1980.
Oculocerebrocutaneous syndrome is a condition characterized by orbital cysts, microphthalmia, porencephaly, agenesis of the corpus callosum, and facial skin tags.
Genitopatellar syndrome is a rare disorder consisting of congenital flexion contractures of the lower extremities, abnormal or missing patellae, and urogenital anomalies. Additional symptoms include microcephaly, severe psychomotor disability. In 2012, it was shown that mutations in the gene KAT6B cause the syndrome. Genitopatellar syndrome (GTPTS) can be caused by heterozygous mutation in the KAT6B gene on chromosome 10q22. The Say-Barber-Biesecker variant of Ohdo syndrome, which has many overlapping features with GTPTS, can also be caused by heterozygous mutation in the KAT6B gene.
13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:
L1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum dysgenesis, spastic paraplegia 1, MASA syndrome, and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS). It is also called L1CAM syndrome and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation, adducted thumbs, spasticity, and hydrocephalus.
Andermann syndrome, also known as agenesis of corpus callosum with neuronopathy (ACCPN) and Charlevoix disease, among other names, is a very rare neurodegenerative genetic disorder that damages the nerves used to control muscles and related to sensation and is often associated with agenesis of the corpus collosum.
Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.
Proud syndrome is a very rare genetic disorder which is characterized by severe intellectual disabilities, corpus callosum agenesis, epilepsy, and spasticity. It is a type of syndromic X-linked intellectual disability.

X-linked complicated corpus callosum dysgenesis is a genetic disorder characterized by dysplasia, hypoplasia or agenesis of the corpus callosum alongside variable intellectual disability and spastic paraplegia. Only 13 cases have been described in medical literature. Transmission is X-linked recessive. It is the mildest subtype of L1 syndrome.
