
A genetic disorder or genetic brain injury (GBI), is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Tietz syndrome, also called Tietz albinism-deafness syndrome or albinism and deafness of Tietz, is an autosomal dominant congenital disorder characterized by deafness and leucism. It is caused by a mutation in the microphthalmia-associated transcription factor (MITF) gene. Tietz syndrome was first described in 1963 by Walter Tietz (1927–2003) a German Physician working in California.

Aniridia is the absence of the iris, a muscular structure that opens and closes the pupil to allow light into the eye. It is also responsible for eye color. Without it the central eye appears all black. It can be congenital, in which both eyes are usually involved, or caused by a penetrant injury. Isolated aniridia is a congenital disorder which is not limited to a defect in iris development, but is a panocular condition with macular and optic nerve hypoplasia, cataract, and corneal changes. Vision may be severely compromised and the disorder is frequently associated with a number of ocular complications: nystagmus, amblyopia, buphthalmos, and cataract. Aniridia in some individuals occurs as part of a syndrome, such as WAGR syndrome, or Gillespie syndrome.

X-linked recessive inheritance is a mode of inheritance in which a mutation in a gene on the X chromosome causes the phenotype to be always expressed in males and in females who are homozygous for the gene mutation, see zygosity. Females with one copy of the mutated gene are carriers.
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections.
Spondyloperipheral dysplasia is an autosomal dominant disorder of bone growth. The condition is characterized by flattened bones of the spine (platyspondyly) and unusually short fingers and toes (brachydactyly). Some affected individuals also have other skeletal abnormalities, short stature, nearsightedness (myopia), hearing loss, and mental retardation. Spondyloperipheral dysplasia is a subtype of collagenopathy, types II and XI.

Cutis laxa or pachydermatocele is a group of rare connective tissue disorders in which the skin becomes inelastic and hangs loosely in folds.
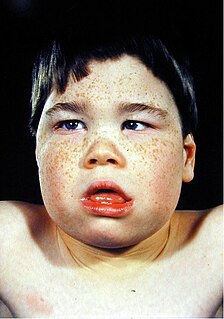
Alpha-thalassemia mental retardation syndrome (ATRX), also called alpha-thalassemia X-linked intellectual disability syndrome, nondeletion type or ATR-X syndrome, is an X-linked recessive condition associated with a mutation in the ATRX gene. Males with this condition tend to be moderately intellectually disabled and have physical characteristics including coarse facial features, microcephaly, hypertelorism, a depressed nasal bridge, a tented upper lip and an everted lower lip. Mild or moderate anemia, associated with alpha-thalassemia, is part of the condition. Females with this mutated gene have no specific signs or features, but if they do, they may demonstrate skewed X chromosome inactivation.

Keutel syndrome (KS) is a rare autosomal recessive genetic disorder characterized by abnormal diffuse cartilage calcification, hypoplasia of the mid-face, peripheral pulmonary stenosis, hearing loss, short distal phalanges (tips) of the fingers and mild mental retardation. Individuals with KS often present with peripheral pulmonary stenosis, brachytelephalangism, sloping forehead, midface hypoplasia, and receding chin. It is associated with abnormalities in the gene coding for matrix gla protein (MGP). Being an autosomal recessive disorder, it may be inherited from two unaffected, abnormal MGP-carrying parents. Thus, people who inherit two affected MGP genes will likely inherit KS.

Woodhouse–Sakati syndrome, is a rare autosomal recessive multisystem disorder which causes malformations throughout the body, and deficiencies affecting the endocrine system.
FG syndrome (FGS) is a rare genetic syndrome caused by one or more recessive genes located on the X chromosome and causing physical anomalies and developmental delays. FG syndrome was named after the first letters of the surnames of the first patients noted with the disease. First reported by American geneticists John M. Opitz and Elisabeth G. Kaveggia in 1974, its major clinical features include intellectual disability, hyperactivity, hypotonia, and a characteristic facial appearance including macrocephaly.
Acropectoral syndrome is an autosomal dominant skeletal dysplasia syndrome affecting the hands, feet, sternum, and lumbosacral spine. A recently proposed candidate gene for preaxial polydactyly is LMBR1, encoding a novel transmembrane receptor, which may be an upstream regulator of SHH. The LMBR1 gene is on human chromosome 7q36.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
Cooks syndrome is a hereditary disorder which is characterized in the hands by bilateral nail hypoplasia on the thumb, index finger, and middle finger, absence of fingernails (anonychia) on the ring finger and little finger, lengthening of the thumbs, and bulbousness of the fingers. In the feet, it is characterized by absence of toenails and absence/hypoplasia of the distal phalanges. In the second study of this disorder, it was found that the intermediate phalanges, proximal phalanges, and metacarpals were unaffected.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.

Mohr–Tranebjærg syndrome (MTS) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. It is characterized by clinical manifestations commencing with early childhood onset hearing loss, followed by adolescent onset progressive or , visual impairment from early adulthood onwards and dementia from the 4th decade onwards. The severity of the symptoms may vary, but they progress usually to severe deafness and dystonia and sometimes are accompanied by cortical deterioration of vision and mental deterioration.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.

Ectrodactyly, split hand, or cleft hand involves the deficiency or absence of one or more central digits of the hand or foot and is also known as split hand/split foot malformation (SHFM). The hands and feet of people with ectrodactyly (ectrodactyls) are often described as "claw-like" and may include only the thumb and one finger with similar abnormalities of the feet.

Brachydactyly type D, also known as short thumb or stub thumb and inaccurately referred to as clubbed thumb, is a condition clinically recognised by a thumb being relatively short and round with an accompanying wider nail bed. The distal phalanx of affected thumbs is approximately two-thirds the length of full-length thumbs. It is the most common type of brachydactyly, or shortness of digits, affecting approximately 2-3% of the population, and is associated with the HOXD13 gene, located on chromosome 2q31.1
White–Sutton syndrome (WHSUS) is a rare neurodevelopmental disorder that affects different systems of the human body. It is mainly characterized by developmental delay, intellectual disability, craniofacial abnormalities and commonly features of autism spectrum disorder (ASD).


