Dementia is a syndrome associated with many neurodegenerative diseases, which is characterized by a general decline in cognitive abilities that impacts a person's ability to perform everyday activities. This typically involves problems with memory, thinking, behavior, and motor control. Aside from memory impairment and a disruption in thought patterns, the most common symptoms include emotional problems, difficulties with language, and decreased motivation. The symptoms may be described as occurring in a continuum over several stages. Dementia ultimately has a significant effect on the individual, caregivers, and on social relationships in general. A diagnosis of dementia requires the observation of a change from a person's usual mental functioning and a greater cognitive decline than what is caused by normal aging.

The temporal lobe is one of the four major lobes of the cerebral cortex in the brain of mammals. The temporal lobe is located beneath the lateral fissure on both cerebral hemispheres of the mammalian brain.

Brodmann area 38, also BA38 or temporopolar area 38 (H), is part of the temporal cortex in the human brain. BA 38 is at the anterior end of the temporal lobe, known as the temporal pole.

Progressive supranuclear palsy (PSP) is a late-onset neurodegenerative disease involving the gradual deterioration and death of specific volumes of the brain. The condition leads to symptoms including loss of balance, slowing of movement, difficulty moving the eyes, and cognitive impairment. PSP may be mistaken for other types of neurodegeneration such as Parkinson's disease, frontotemporal dementia and Alzheimer's disease. The cause of the condition is uncertain, but involves the accumulation of tau protein within the brain. Medications such as levodopa and amantadine may be useful in some cases.

Frontotemporal lobar degeneration (FTLD) is a pathological process that occurs in frontotemporal dementia. It is characterized by atrophy in the frontal lobe and temporal lobe of the brain, with sparing of the parietal and occipital lobes.
Semantic dementia (SD), also known as semantic variant primary progressive aphasia (svPPA), is a progressive neurodegenerative disorder characterized by loss of semantic memory in both the verbal and non-verbal domains. However, the most common presenting symptoms are in the verbal domain. Semantic dementia is a disorder of semantic memory that causes patients to lose the ability to match words or images to their meanings. However, it is fairly rare for patients with semantic dementia to develop category specific impairments, though there have been documented cases of it occurring. Typically, a more generalized semantic impairment results from dimmed semantic representations in the brain.
Progressive nonfluent aphasia (PNFA) is one of three clinical syndromes associated with frontotemporal lobar degeneration. PNFA has an insidious onset of language deficits over time as opposed to other stroke-based aphasias, which occur acutely following trauma to the brain. The specific degeneration of the frontal and temporal lobes in PNFA creates hallmark language deficits differentiating this disorder from other Alzheimer-type disorders by the initial absence of other cognitive and memory deficits. This disorder commonly has a primary effect on the left hemisphere, causing the symptomatic display of expressive language deficits and sometimes may disrupt receptive abilities in comprehending grammatically complex language.

Primary progressive aphasia (PPA) is a type of neurological syndrome in which language capabilities slowly and progressively become impaired. As with other types of aphasia, the symptoms that accompany PPA depend on what parts of the left hemisphere are significantly damaged. However, unlike most other aphasias, PPA results from continuous deterioration in brain tissue, which leads to early symptoms being far less detrimental than later symptoms.

Hippocampal sclerosis (HS) or mesial temporal sclerosis (MTS) is a neuropathological condition with severe neuronal cell loss and gliosis in the hippocampus. Neuroimaging tests such as magnetic resonance imaging (MRI) and positron emission tomography (PET) may identify individuals with hippocampal sclerosis. Hippocampal sclerosis occurs in 3 distinct settings: mesial temporal lobe epilepsy, adult neurodegenerative disease and acute brain injury.

Corticobasal degeneration (CBD) is a rare neurodegenerative disease involving the cerebral cortex and the basal ganglia. CBD symptoms typically begin in people from 50 to 70 years of age, and typical survival before death is eight years. It is characterized by marked disorders in movement and cognition, and is classified as one of the Parkinson plus syndromes. Diagnosis is difficult, as symptoms are often similar to those of other disorders, such as Parkinson's disease, progressive supranuclear palsy, and dementia with Lewy bodies, and a definitive diagnosis of CBD can only be made upon neuropathologic examination.
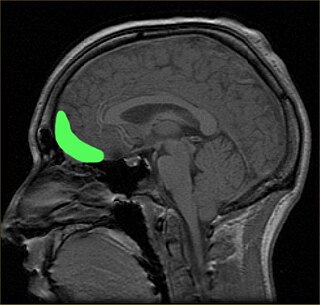
The orbitofrontal cortex (OFC) is a prefrontal cortex region in the frontal lobes of the brain which is involved in the cognitive process of decision-making. In non-human primates it consists of the association cortex areas Brodmann area 11, 12 and 13; in humans it consists of Brodmann area 10, 11 and 47.

Frontal lobe disorder, also frontal lobe syndrome, is an impairment of the frontal lobe of the brain due to disease or frontal lobe injury. The frontal lobe plays a key role in executive functions such as motivation, planning, social behaviour, and speech production. Frontal lobe syndrome can be caused by a range of conditions including head trauma, tumours, neurodegenerative diseases, neurodevelopmental disorders, neurosurgery and cerebrovascular disease. Frontal lobe impairment can be detected by recognition of typical signs and symptoms, use of simple screening tests, and specialist neurological testing.

Tauopathies are a class of neurodegenerative diseases characterized by the aggregation of abnormal tau protein. Hyperphosphorylation of tau proteins causes them to dissociate from microtubules and form insoluble aggregates called neurofibrillary tangles. Various neuropathologic phenotypes have been described based on the anatomical regions and cell types involved as well as the unique tau isoforms making up these deposits. The designation 'primary tauopathy' is assigned to disorders where the predominant feature is the deposition of tau protein. Alternatively, diseases exhibiting tau pathologies attributed to different and varied underlying causes are termed 'secondary tauopathies'. Some neuropathologic phenotypes involving tau protein is Alzheimer's disease, Pick disease, Progressive supranuclear palsy, and corticobasal degeneration.

Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) is an autosomal dominant neurodegenerative tauopathy and Parkinson plus syndrome. FTDP-17 is caused by mutations in the MAPT gene located on the q arm of chromosome 17, and has three cardinal features: behavioral and personality changes, cognitive impairment, and motor symptoms. FTDP-17 was defined during the International Consensus Conference in Ann Arbor, Michigan, in 1996.

Transmembrane protein 106B is a protein that is encoded by the TMEM106B gene. It is found primarily within neurons and oligodendrocytes in the central nervous system with its subcellular location being in lysosomal membranes. TMEM106B helps facilitate important functions for maintaining a healthy lysosome, and therefore certain mutations and polymorphisms can lead to issues with proper lysosomal function. Lysosomes are in charge of clearing out mis-folded proteins and other debris, and thus, play an important role in neurodegenerative diseases that are driven by the accumulation of various mis-folded proteins and aggregates. Due to its impact on lysosomal function, TMEM106B has been investigated and found to be associated to multiple neurodegenerative diseases.
The applause sign is a behavioural indicator, relevant to neurodegenerative conditions, characterised by a patient’s inability to execute the same number of hand claps as demonstrated by an examiner.

C9orf72 is a protein which in humans is encoded by the gene C9orf72.
Corticobasal syndrome (CBS) is a rare, progressive atypical Parkinsonism syndrome and is a tauopathy related to frontotemporal dementia. CBS is typically caused by the deposit of tau proteins forming in different areas of the brain.

LATE is a term that describes a prevalent condition with impaired memory and thinking in advanced age, often culminating in the dementia clinical syndrome. In other words, the symptoms of LATE are similar to those of Alzheimer's disease.
The Cambridge Behavioural Inventory (CBI) and its revised version, Cambridge Behavioural Inventory-Revised (CBI-R), are informant-based questionnaires that evaluate the emergence of behavioural symptoms in neurodegenerative brain disorders, including Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), and frontotemporal dementia (FTD).