Fragile X syndrome (FXS) is a genetic disorder characterized by mild-to-moderate intellectual disability. The average IQ in males with FXS is under 55, while about two thirds of affected females are intellectually disabled. Physical features may include a long and narrow face, large ears, flexible fingers, and large testicles. About a third of those affected have features of autism such as problems with social interactions and delayed speech. Hyperactivity is common, and seizures occur in about 10%. Males are usually more affected than females.
Macroorchidism is a disorder found in males, specifically in children, where a subject has abnormally large testes. The condition is commonly inherited in connection with fragile X syndrome (FXS), which is also the second most common genetic cause of intellectual disability. The condition is also a rare sign of the McCune-Albright syndrome. The opposite of macroorchidism is called microorchidism, which is the condition of abnormally small testes.
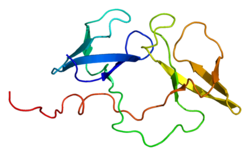
RNA-binding proteins are proteins that bind to the double or single stranded RNA in cells and participate in forming ribonucleoprotein complexes. RBPs contain various structural motifs, such as RNA recognition motif (RRM), dsRNA binding domain, zinc finger and others. They are cytoplasmic and nuclear proteins. However, since most mature RNA is exported from the nucleus relatively quickly, most RBPs in the nucleus exist as complexes of protein and pre-mRNA called heterogeneous ribonucleoprotein particles (hnRNPs). RBPs have crucial roles in various cellular processes such as: cellular function, transport and localization. They especially play a major role in post-transcriptional control of RNAs, such as: splicing, polyadenylation, mRNA stabilization, mRNA localization and translation. Eukaryotic cells express diverse RBPs with unique RNA-binding activity and protein–protein interaction. According to the Eukaryotic RBP Database (EuRBPDB), there are 2961 genes encoding RBPs in humans. During evolution, the diversity of RBPs greatly increased with the increase in the number of introns. Diversity enabled eukaryotic cells to utilize RNA exons in various arrangements, giving rise to a unique RNP (ribonucleoprotein) for each RNA. Although RBPs have a crucial role in post-transcriptional regulation in gene expression, relatively few RBPs have been studied systematically.It has now become clear that RNA–RBP interactions play important roles in many biological processes among organisms.
A trinucleotide repeat expansion, also known as a triplet repeat expansion, is the DNA mutation responsible for causing any type of disorder categorized as a trinucleotide repeat disorder. These are labelled in dynamical genetics as dynamic mutations. Triplet expansion is caused by slippage during DNA replication, also known as "copy choice" DNA replication. Due to the repetitive nature of the DNA sequence in these regions, 'loop out' structures may form during DNA replication while maintaining complementary base pairing between the parent strand and daughter strand being synthesized. If the loop out structure is formed from the sequence on the daughter strand this will result in an increase in the number of repeats. However, if the loop out structure is formed on the parent strand, a decrease in the number of repeats occurs. It appears that expansion of these repeats is more common than reduction. Generally, the larger the expansion the more likely they are to cause disease or increase the severity of disease. Other proposed mechanisms for expansion and reduction involve the interaction of RNA and DNA molecules.

Neurexins (NRXN) are a family of presynaptic cell adhesion proteins that have roles in connecting neurons at the synapse. They are located mostly on the presynaptic membrane and contain a single transmembrane domain. The extracellular domain interacts with proteins in the synaptic cleft, most notably neuroligin, while the intracellular cytoplasmic portion interacts with proteins associated with exocytosis. Neurexin and neuroligin "shake hands," resulting in the connection between the two neurons and the production of a synapse. Neurexins mediate signaling across the synapse, and influence the properties of neural networks by synapse specificity. Neurexins were discovered as receptors for α-latrotoxin, a vertebrate-specific toxin in black widow spider venom that binds to presynaptic receptors and induces massive neurotransmitter release. In humans, alterations in genes encoding neurexins are implicated in autism and other cognitive diseases, such as Tourette syndrome and schizophrenia.

Glutamate ionotropic receptor kainate type subunit 2, also known as ionotropic glutamate receptor 6 or GluR6, is a protein that in humans is encoded by the GRIK2 gene.

Fragile X mental retardation syndrome-related protein 1 is a protein that in humans is encoded by the FXR1 gene.

Glutamate receptor, ionotropic, kainate 1, also known as GRIK1, is a protein that in humans is encoded by the GRIK1 gene.

Fragile X mental retardation syndrome-related protein 2 is a protein that in humans is encoded by the FXR2 gene.

AF4/FMR2 family member 2 is a protein that in humans is encoded by the AFF2 gene. Mutations in AFF2 are implicated in cases of breast cancer.

CGG triplet repeat-binding protein 1 is a protein that in humans is encoded by the CGGBP1 gene.

Nuclear fragile X mental retardation-interacting protein 1 is a protein that in humans is encoded by the NUFIP1 gene.
Activity-dependent plasticity is a form of functional and structural neuroplasticity that arises from the use of cognitive functions and personal experience; hence, it is the biological basis for learning and the formation of new memories. Activity-dependent plasticity is a form of neuroplasticity that arises from intrinsic or endogenous activity, as opposed to forms of neuroplasticity that arise from extrinsic or exogenous factors, such as electrical brain stimulation- or drug-induced neuroplasticity. The brain's ability to remodel itself forms the basis of the brain's capacity to retain memories, improve motor function, and enhance comprehension and speech amongst other things. It is this trait to retain and form memories that is associated with neural plasticity and therefore many of the functions individuals perform on a daily basis. This plasticity occurs as a result of changes in gene expression which are triggered by signaling cascades that are activated by various signaling molecules during increased neuronal activity.

Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder most frequently seen in male premutation carriers of Fragile X syndrome (FXS) over the age of 50. The main clinical features of FXTAS include problems of movement with cerebellar gait ataxia and action tremor. Associated features include parkinsonism, cognitive decline, and dysfunction of the autonomic nervous system. FXTAS is found in Fragile X "premutation" carriers, which is defined as a trinucleotide repeat expansion of 55-200 CGG repeats in the Fragile X mental retardation-1 (FMR1) gene. 4-40 CGG repeats in this gene is considered normal, while individual with >200 repeats have full Fragile X Syndrome.

In molecular biology, bantam microRNA is a short RNA molecule. MicroRNAs function to regulate the expression levels of other genes by several mechanisms.
In molecular biology, FMR1 antisense RNA 1 (FMR1-AS1), also known as ASFMR1 or FMR4, is a long non-coding RNA. The FMR1-AS1 gene overlaps, and is antisense to, the CGG repeat region of the FMR1 gene. Its expression is upregulated in fragile X syndrome premutation carriers, and silenced in patients with fragile X syndrome. FMR1-AS1 has an anti-apoptotic function.
Fragile X-associated Primary Ovarian Insufficiency (FXPOI) is the most common genetic cause of premature ovarian failure in women with a normal karyotype 46, XX. The expansion of a CGG repeat in the 5' untranslated region of the FMR1 gene from the normal range of 5-45 repeats to the premutation range of 55-199 CGGs leads to risk of FXPOI for ovary-bearing individuals. About 1:150-1:200 women in the US population carry a premutation. Women who carry an FMR1 premutation have a roughly 20% risk of being diagnosed with FXPOI, compared to 1% for the general population, and an 8-15% risk of developing the neurogenerative tremor/ataxia disorder (FXTAS). FMR1 premutation women are also at increased risk of having a child with a CGG repeat that is expanded to >200 repeats. Individuals with a full mutation, unlike the premutation, produce little to no mRNA or protein from the FMR1 gene and are affected with Fragile X syndrome.
Nagwa Abdel Meguid is an Egyptian geneticist and 2002 winner of the L’Oreal UNESCO Award for Women in Science for Africa and the Middle East. Her research has "identified several genetic mutations that cause common syndromes such as the fragile X syndrome and Autism".
David L. Nelson is an American human geneticist, currently an associate director at the Intellectual and Developmental Disabilities Research Center (1995), and professor at the Department of Molecular and Human Genetics at Baylor College of Medicine BCM since 1999. Since 2018, he is the director at the Cancer and Cell Biology Ph.D program, and the director of Integrative Molecular and Biomedical Sciences Ph.D since 2015 at BCM.
Stephen T. Warren was an American geneticist and academic. He was the William Patterson Timmie Professor of Human Genetics and the Charles Howard Candler Chair of Human Genetics. He was the former Founding Chairman of the Department of Human Genetics at Emory University School of Medicine. He was an Investigator with the Howard Hughes Medical Institute from 1991 until 2002, when he resigned to found the Human Genetics department. Warren is well known for his work in the field of Human Genetics. His research was focused on the mechanistic understanding of fragile X syndrome, a leading cause of inherited developmental disability and autism. In 2020, Warren stepped down as department chair after 20 years in that position.