
Abetalipoproteinemia is a disorder characterized by abnormal absorption of fat and fat-soluble vitamins from food. It is caused by a mutation in microsomal triglyceride transfer protein resulting in deficiencies in the apolipoproteins B-48 and B-100, which are used in the synthesis and exportation of chylomicrons and VLDL respectively. It is not to be confused with familial dysbetalipoproteinemia.
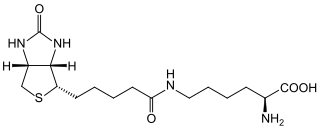
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.
Vitamin E deficiency in humans is a very rare condition, occurring as a consequence of abnormalities in dietary fat absorption or metabolism rather than from a diet low in vitamin E. Collectively the EARs, RDAs, AIs and ULs for vitamin E and other essential nutrients are referred to as Dietary Reference Intakes (DRIs). Vitamin E deficiency can cause nerve problems due to poor conduction of electrical impulses along nerves due to changes in nerve membrane structure and function.
Lecithin cholesterol acyltransferase deficiency is a disorder of lipoprotein metabolism. The disease has two forms: Familial LCAT deficiency, in which there is complete LCAT deficiency, and Fish-eye disease, in which there is a partial deficiency.

Lipoprotein lipase deficiency is a genetic disorder in which a person has a defective gene for lipoprotein lipase, which leads to very high triglycerides, which in turn causes stomach pain and deposits of fat under the skin, and which can lead to problems with the pancreas and liver, which in turn can lead to diabetes. The disorder only occurs if a child acquires the defective gene from both parents. It is managed by restricting fat in diet to less than 20 g/day.

Arterial tortuosity syndrome is an extremely rare congenital connective tissue condition disorder characterized by tortuosity, elongation, stenosis, or aneurysms in major and medium-size arteries including the aorta. It is associated with hyperextensible skin and hypermobility of joints, however symptoms vary depending on the person. Because ATS is so rare, relatively little is known about the disease compared to more common diseases.

GLUT1 deficiency syndrome, also known as GLUT1-DS, De Vivo disease or Glucose transporter type 1 deficiency syndrome, is an autosomal dominant genetic metabolic disorder associated with a deficiency of GLUT1, the protein that transports glucose across the blood brain barrier. Glucose Transporter Type 1 Deficiency Syndrome has an estimated birth incidence of 1 in 90,000 to 1 in 24,300. This birth incidence translates to an estimated prevalence of 3,000 to 7,000 in the U.S.
MT-ATP6 is a mitochondrial gene with the full name 'mitochondrially encoded ATP synthase membrane subunit 6' that encodes the ATP synthase Fo subunit 6. This subunit belongs to the Fo complex of the large, transmembrane F-type ATP synthase. This enzyme, which is also known as complex V, is responsible for the final step of oxidative phosphorylation in the electron transport chain. Specifically, one segment of ATP synthase allows positively charged ions, called protons, to flow across a specialized membrane inside mitochondria. Another segment of the enzyme uses the energy created by this proton flow to convert a molecule called adenosine diphosphate (ADP) to ATP. Mutations in the MT-ATP6 gene have been found in approximately 10 to 20 percent of people with Leigh syndrome.

Cav2.1, also called the P/Q voltage-dependent calcium channel, is a calcium channel found mainly in the brain. Specifically, it is found on the presynaptic terminals of neurons in the brain and cerebellum. Cav2.1 plays an important role in controlling the release of neurotransmitters between neurons. It is composed of multiple subunits, including alpha-1, beta, alpha-2/delta, and gamma subunits. The alpha-1 subunit is the pore-forming subunit, meaning that the calcium ions flow through it. Different kinds of calcium channels have different isoforms (versions) of the alpha-1 subunit. Cav2.1 has the alpha-1A subunit, which is encoded by the CACNA1A gene. Mutations in CACNA1A have been associated with various neurologic disorders, including familial hemiplegic migraine, episodic ataxia type 2, and spinocerebellar ataxia type 6.

Cytochrome c oxidase II is a protein in eukaryotes that is encoded by the MT-CO2 gene. Cytochrome c oxidase subunit II, abbreviated COXII, COX2, COII, or MT-CO2, is the second subunit of cytochrome c oxidase. It is also one of the three mitochondrial DNA (mtDNA) encoded subunits of respiratory complex IV.

Alpha-tocopherol transfer protein (α-TTP) is a protein that in humans is encoded by the TTPA gene.
Mitochondrially encoded tRNA phenylalanine also known as MT-TF is a transfer RNA which in humans is encoded by the mitochondrial MT-TF gene.
Mitochondrially encoded tRNA glycine also known as MT-TG is a transfer RNA which in humans is encoded by the mitochondrial MT-TG gene.
Mitochondrially encoded tRNA isoleucine also known as MT-TI is a transfer RNA which in humans is encoded by the mitochondrial MT-TI gene.
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.
Mitochondrially encoded tRNA asparagine also known as MT-TN is a transfer RNA which in humans is encoded by the mitochondrial MT-TN gene.
Mitochondrially encoded tRNA proline also known as MT-TP is a transfer RNA that in humans is encoded by the mitochondrial MT-TP gene.
Mitochondrially encoded tRNA tyrosine, also known as MT-TY, is a transfer RNA which in humans is encoded by the mitochondrial MT-TY gene.

Dopamine beta (β)-hydroxylase deficiency is a human medical condition involving inadequate dopamine beta-hydroxylase. It is characterized by increased amounts of serum dopamine and the absence of norepinephrine (NE) and epinephrine.
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare genetic disease. It is a primary immunodeficiency featured by molecular defects in IL12/IFNγ dependent signalling pathway, leading to increased susceptibility to local or disseminated infections by environmental mycobacteria, Mycobacterium bovis Bacille Calmette-Guerin strain, nontyphoidal and typhoidal Salmonella serotypes.
