
Biotin (or vitamin B7) is one of the B vitamins. It is involved in a wide range of metabolic processes, both in humans and in other organisms, primarily related to the utilization of fats, carbohydrates, and amino acids. The name biotin derives from the Greek word "bios" (to live) and the suffix "-in" (a general chemical suffix used in organic chemistry).

Carnitine is a quaternary ammonium compound involved in metabolism in most mammals, plants, and some bacteria. In support of energy metabolism, carnitine transports long-chain fatty acids into mitochondria to be oxidized for energy production, and also participates in removing products of metabolism from cells. Given its key metabolic roles, carnitine is concentrated in tissues like skeletal and cardiac muscle that metabolize fatty acids as an energy source. Generally individuals, including strict vegetarians, synthesize enough L-carnitine in vivo.
Encephalopathy means any disorder or disease of the brain, especially chronic degenerative conditions. In modern usage, encephalopathy does not refer to a single disease, but rather to a syndrome of overall brain dysfunction; this syndrome has many possible organic and inorganic causes.
Propionic acidemia, also known as propionic aciduria or propionyl-CoA carboxylase deficiency, is a rare autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia.
Hypotonia is a state of low muscle tone, often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength. Hypotonia is a lack of resistance to passive movement, whereas muscle weakness results in impaired active movement. Central hypotonia originates from the central nervous system, while peripheral hypotonia is related to problems within the spinal cord, peripheral nerves and/or skeletal muscles. Severe hypotonia in infancy is commonly known as floppy baby syndrome. Recognizing hypotonia, even in early infancy, is usually relatively straightforward, but diagnosing the underlying cause can be difficult and often unsuccessful. The long-term effects of hypotonia on a child's development and later life depend primarily on the severity of the muscle weakness and the nature of the cause. Some disorders have a specific treatment but the principal treatment for most hypotonia of idiopathic or neurologic cause is physical therapy and/or occupational therapy for remediation.

Citrullinemia is an autosomal recessive urea cycle disorder that causes ammonia and other toxic substances to accumulate in the blood.
Holocarboxylase synthetase deficiency is an inherited metabolic disorder in which the body is unable to use the vitamin biotin effectively. This disorder is classified as a multiple carboxylase deficiency, a group of disorders characterized by impaired activity of certain enzymes that depend on biotin. Symptoms are very similar to biotinidase deficiency and treatment – large doses of biotin – is also the same.
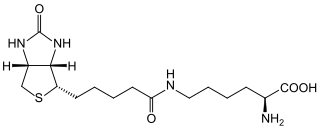
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.
Holocarboxylase synthetase ), also known as protein—biotin ligase, is a family of enzymes. This enzyme is important for the effective use of biotin, a B vitamin found in foods such as liver, egg yolks, and milk. In many of the body's tissues, holocarboxylase synthetase activates other specific enzymes by attaching biotin to them. These carboxylases are involved in many critical cellular functions, including the production and breakdown of proteins, fats, and carbohydrates.

3-Methylcrotonyl-CoA carboxylase deficiency also known as 3-Methylcrotonylglycinuria or BMCC deficiency is an inherited disorder in which the body is unable to process certain proteins properly. People with this disorder have inadequate levels of an enzyme that helps break down proteins containing the amino acid leucine. This condition affects an estimated 1 in 50,000 individuals worldwide.
Carbamoyl phosphate synthetase I deficiency is an autosomal recessive metabolic disorder that causes ammonia to accumulate in the blood due to a lack of the enzyme carbamoyl phosphate synthetase I. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
Pyruvate carboxylase deficiency is an inherited disorder that causes lactic acid to accumulate in the blood. High levels of these substances can damage the body's organs and tissues, particularly in the nervous system. Pyruvate carboxylase deficiency is a rare condition, with an estimated incidence of 1 in 250,000 births worldwide. Type A of the disease appears to be much more common in some Algonkian Indian tribes in eastern Canada, while the type B disease is more present in European populations.
Glutathione synthetase deficiency (GSD) is a rare autosomal recessive metabolic disorder that prevents the production of glutathione. Glutathione helps prevent damage to cells by neutralizing harmful molecules generated during energy production. Glutathione also plays a role in processing medications and cancer-causing compounds (carcinogens), and building DNA, proteins, and other important cellular components.
Biotinidase, also known as biotinase, is an enzyme that in humans is encoded by the BTD gene.

Carbamoyl phosphate synthetase catalyzes the ATP-dependent synthesis of carbamoyl phosphate from glutamine or ammonia and bicarbonate. This enzyme catalyzes the reaction of ATP and bicarbonate to produce carboxy phosphate and ADP. Carboxy phosphate reacts with ammonia to give carbamic acid. In turn, carbamic acid reacts with a second ATP to give carbamoyl phosphate plus ADP.
Biotin deficiency is a nutritional disorder which can become serious, even fatal, if allowed to progress untreated. It can occur in people of any age, ancestry, or of either gender. Biotin is part of the B vitamin family. Biotin deficiency rarely occurs among healthy people because the daily requirement of biotin is low, many foods provide adequate amounts of it, intestinal bacteria synthesize small amounts of it, and the body effectively scavenges and recycles it in the kidneys during production of urine. However, deficiencies can be caused by consuming raw egg whites over a period of weeks to months. Egg whites contain high levels of avidin, a protein that binds biotin strongly. When cooked, avidin is partially denatured and binding to biotin is reduced. However one study showed that 30-40% of the avidin activity was still present in the white after frying or boiling. Genetic disorders such as biotinidase deficiency, multiple carboxylase deficiency, and holocarboxylase synthetase deficiency can also lead to inborn or late-onset forms of biotin deficiency. In all cases – dietary, genetic, or otherwise – supplementation with biotin is the primary method of treatment.

Citrullinemia type I (CTLN1), also known as arginosuccinate synthetase deficiency, is a rare disease caused by a deficiency in argininosuccinate synthetase, an enzyme involved in excreting excess nitrogen from the body. There are mild and severe forms of the disease, which is one of the urea cycle disorders.