
Crystallography is the experimental science of determining the arrangement of atoms in crystalline solids. Crystallography is a fundamental subject in the fields of materials science and solid-state physics. The word "crystallography" is derived from the Greek words κρύσταλλος (krystallos) "clear ice, rock-crystal", with its meaning extending to all solids with some degree of transparency, and γράφειν (graphein) "to write". In July 2012, the United Nations recognised the importance of the science of crystallography by proclaiming that 2014 would be the International Year of Crystallography.

X-ray crystallography is the experimental science determining the atomic and molecular structure of a crystal, in which the crystalline structure causes a beam of incident X-rays to diffract into many specific directions. By measuring the angles and intensities of these diffracted beams, a crystallographer can produce a three-dimensional picture of the density of electrons within the crystal. From this electron density, the mean positions of the atoms in the crystal can be determined, as well as their chemical bonds, their crystallographic disorder, and various other information.

X-ray scattering techniques are a family of non-destructive analytical techniques which reveal information about the crystal structure, chemical composition, and physical properties of materials and thin films. These techniques are based on observing the scattered intensity of an X-ray beam hitting a sample as a function of incident and scattered angle, polarization, and wavelength or energy.

Electron diffraction refers to the bending of electron beams around atomic structures. This behaviour, typical for waves, is applicable to electrons due to the wave–particle duality stating that electrons behave as both particles and waves. Since the diffracted beams interfere, they generate diffraction patterns widely used for analysis of the objects which caused the diffraction. Therefore, electron diffraction can also refer to derived experimental techniques used for material characterization. This technique is similar to X-ray and neutron diffraction.

Miller indices form a notation system in crystallography for lattice planes in crystal (Bravais) lattices.
In physics, the phase problem is the problem of loss of information concerning the phase that can occur when making a physical measurement. The name comes from the field of X-ray crystallography, where the phase problem has to be solved for the determination of a structure from diffraction data. The phase problem is also met in the fields of imaging and signal processing. Various approaches of phase retrieval have been developed over the years.
Rietveld refinement is a technique described by Hugo Rietveld for use in the characterisation of crystalline materials. The neutron and X-ray diffraction of powder samples results in a pattern characterised by reflections at certain positions. The height, width and position of these reflections can be used to determine many aspects of the material's structure.
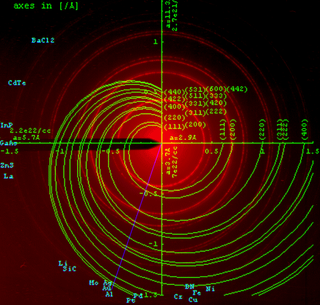
Powder diffraction is a scientific technique using X-ray, neutron, or electron diffraction on powder or microcrystalline samples for structural characterization of materials. An instrument dedicated to performing such powder measurements is called a powder diffractometer.
The Debye–Waller factor (DWF), named after Peter Debye and Ivar Waller, is used in condensed matter physics to describe the attenuation of x-ray scattering or coherent neutron scattering caused by thermal motion. It is also called the B factor, atomic B factor, or temperature factor. Often, "Debye–Waller factor" is used as a generic term that comprises the Lamb–Mössbauer factor of incoherent neutron scattering and Mössbauer spectroscopy.
In condensed matter physics and crystallography, the static structure factor is a mathematical description of how a material scatters incident radiation. The structure factor is a critical tool in the interpretation of scattering patterns obtained in X-ray, electron and neutron diffraction experiments.

Fiber diffraction is a subarea of scattering, an area in which molecular structure is determined from scattering data. In fiber diffraction the scattering pattern does not change, as the sample is rotated about a unique axis. Such uniaxial symmetry is frequent with filaments or fibers consisting of biological or man-made macromolecules. In crystallography fiber symmetry is an aggravation regarding the determination of crystal structure, because reflexions are smeared and may overlap in the fiber diffraction pattern. Materials science considers fiber symmetry a simplification, because almost the complete obtainable structure information is in a single two-dimensional (2D) diffraction pattern exposed on photographic film or on a 2D detector. 2 instead of 3 co-ordinate directions suffice to describe fiber diffraction.
In crystallography, the R-factor is a measure of the agreement between the crystallographic model and the experimental X-ray diffraction data. In other words, it is a measure of how well the refined structure predicts the observed data. The value is also sometimes called the discrepancy index, as it mathematically describes the difference between the experimental observations and the ideal calculated values. It is defined by the following equation:

Absolute configuration refers to the spatial arrangement of atoms within a chiral molecular entity and its resultant stereochemical description. Absolute configuration is typically relevant in organic molecules, where carbon is bonded to four different substituents. This type of construction creates two possible enantiomers. Absolute configuration uses a set of rules to describe the relative positions of each bond around the chiral center atom. The most common labeling method uses the descriptors R or S is based on the Cahn–Ingold–Prelog priority rules. R and S refer to Rectus and Sinister, which are Latin for right and left, respectively.
Multi-wavelength anomalous diffraction is a technique used in X-ray crystallography that facilitates the determination of the three-dimensional structure of biological macromolecules via solution of the phase problem.
Molecular replacement is a method of solving the phase problem in X-ray crystallography. MR relies upon the existence of a previously solved protein structure which is similar to our unknown structure from which the diffraction data is derived. This could come from a homologous protein, or from the lower-resolution protein NMR structure of the same protein.

Johannes Martin Bijvoet was a Dutch chemist and crystallographer at the van 't Hoff Laboratory at Utrecht University. He is famous for devising a method of establishing the absolute configuration of molecules. In 1946, he became member of the Royal Netherlands Academy of Arts and Sciences.
Anomalous X-ray scattering is a non-destructive determination technique within X-ray diffraction that makes use of the anomalous dispersion that occurs when a wavelength is selected that is in the vicinity of an absorption edge of one of the constituent elements of the sample. It is used in materials research to study nanometer sized differences in structure.
Multiple isomorphous replacement (MIR) is historically the most common approach to solving the phase problem in X-ray crystallography studies of proteins. For protein crystals this method is conducted by soaking the crystal of a sample to be analyzed with a heavy atom solution or co-crystallization with the heavy atom. The addition of the heavy atom to the structure should not affect the crystal formation or unit cell dimensions in comparison to its native form, hence, they should be isomorphic.
Single-wavelength anomalous diffraction (SAD) is a technique used in X-ray crystallography that facilitates the determination of the structure of proteins or other biological macromolecules by allowing the solution of the phase problem. In contrast to multi-wavelength anomalous diffraction, SAD uses a single dataset at a single appropriate wavelength. One advantage of the technique is the minimization of time spent in the beam by the crystal, thus reducing potential radiation damage to the molecule while collecting data. SAD is sometimes called "single-wavelength anomalous dispersion", but no dispersive differences are used in this technique since the data are collected at a single wavelength. Today, selenium-SAD is commonly used for experimental phasing due to the development of methods for selenomethionine incorporation into recombinant proteins.
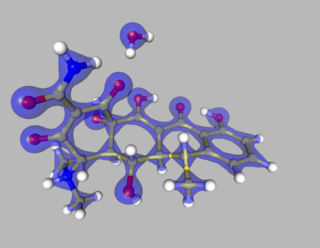
The Multipole Density Formalism is an X-ray crystallography method of electron density modelling proposed by Niels K. Hansen and Philip Coppens in 1978. Unlike the commonly used Independent Atom Model, the Hansen-Coppens Formalism presents an aspherical approach, allowing one to model the electron distribution around a nucleus separately in different directions and therefore describe numerous chemical features of a molecule inside the unit cell of an examined crystal in detail.


