Related Research Articles

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Eosinophilia is a condition in which the eosinophil count in the peripheral blood exceeds 5×108/L (500/μL). Hypereosinophilia is an elevation in an individual's circulating blood eosinophil count above 1.5 × 109/L (i.e. 1,500/μL). The hypereosinophilic syndrome is a sustained elevation in this count above 1.5 × 109/L (i.e. 1,500/μL) that is also associated with evidence of eosinophil-based tissue injury.

Fanconi anemia (FA) is a rare, AR, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.
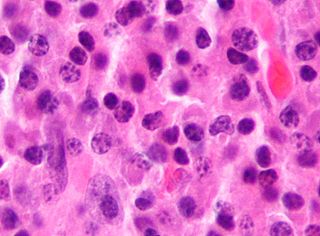
Tumors of the hematopoietic and lymphoid tissues or tumours of the haematopoietic and lymphoid tissues are tumors that affect the blood, bone marrow, lymph, and lymphatic system. Because these tissues are all intimately connected through both the circulatory system and the immune system, a disease affecting one will often affect the others as well, making aplasia, myeloproliferation and lymphoproliferation closely related and often overlapping problems. While uncommon in solid tumors, chromosomal translocations are a common cause of these diseases. This commonly leads to a different approach in diagnosis and treatment of hematological malignancies. Hematological malignancies are malignant neoplasms ("cancer"), and they are generally treated by specialists in hematology and/or oncology. In some centers "hematology/oncology" is a single subspecialty of internal medicine while in others they are considered separate divisions. Not all hematological disorders are malignant ("cancerous"); these other blood conditions may also be managed by a hematologist.
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections. It causes severe pyogenic infections. It can be caused by autosomal dominant inheritance of the ELANE gene, autosomal recessive inheritance of the HAX1 gene. There is an increased risk of leukemia and myelodysplastic cancers.
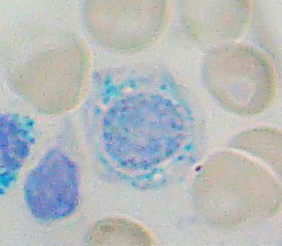
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.

Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype. The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, and MDS/AML, but these components do not always occur. DKC is characterized by short telomeres. Some of the manifestations resemble premature ageing and cognitive impairment can be a feature. The disease initially mainly affects the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.

Acute myeloid leukemia (AML) is a cancer of the myeloid line of blood cells, characterized by the rapid growth of abnormal cells that build up in the bone marrow and blood and interfere with normal blood cell production. Symptoms may include feeling tired, shortness of breath, easy bruising and bleeding, and increased risk of infection. Occasionally, spread may occur to the brain, skin, or gums. As an acute leukemia, AML progresses rapidly, and is typically fatal within weeks or months if left untreated.

GATA-binding factor 1 or GATA-1 is the founding member of the GATA family of transcription factors. This protein is widely expressed throughout vertebrate species. In humans and mice, it is encoded by the GATA1 and Gata1 genes, respectively. These genes are located on the X chromosome in both species.

Chronic myelomonocytic leukemia (CMML) is a type of leukemia, which are cancers of the blood-forming cells of the bone marrow. In adults, blood cells are formed in the bone marrow, by a process that is known as haematopoiesis. In CMML, there are increased numbers of monocytes and immature blood cells (blasts) in the peripheral blood and bone marrow, as well as abnormal looking cells (dysplasia) in at least one type of blood cell.
Juvenile myelomonocytic leukemia (JMML) is a rare form of chronic leukemia that affects children, commonly those aged four and younger. The name JMML now encompasses all diagnoses formerly referred to as juvenile chronic myeloid leukemia (JCML), chronic myelomonocytic leukemia of infancy, and infantile monosomy 7 syndrome. The average age of patients at diagnosis is two (2) years old. The World Health Organization has included JMML as a subcategory of myelodysplastic and myeloproliferative disorders.

ETV6 protein is a transcription factor that in humans is encoded by the ETV6 gene. The ETV6 protein regulates the development and growth of diverse cell types, particularly those of hematological tissues. However, its gene, ETV6 frequently suffers various mutations that lead to an array of potentially lethal cancers, i.e., ETV6 is a clinically significant proto-oncogene in that it can fuse with other genes to drive the development and/or progression of certain cancers. However, ETV6 is also an anti-oncogene or tumor suppressor gene in that mutations in it that encode for a truncated and therefore inactive protein are also associated with certain types of cancers.
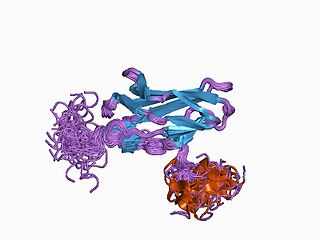
Runt-related transcription factor 1 (RUNX1) also known as acute myeloid leukemia 1 protein (AML1) or core-binding factor subunit alpha-2 (CBFA2) is a protein that in humans is encoded by the RUNX1 gene.

MN1 is a gene found on human chromosome 22, with gene map locus 22q12.3-qter. Its official full name is meningioma 1 because it is disrupted by a balanced translocation (4;22) in a meningioma.

GATA2 or GATA-binding factor 2 is a transcription factor, i.e. a nuclear protein which regulates the expression of genes. It regulates many genes that are critical for the embryonic development, self-renewal, maintenance, and functionality of blood-forming, lympathic system-forming, and other tissue-forming stem cells. GATA2 is encoded by the GATA2 gene, a gene which often suffers germline and somatic mutations which lead to a wide range of familial and sporadic diseases, respectively. The gene and its product are targets for the treatment of these diseases.

Acute megakaryoblastic leukemia (AMKL) is life-threatening leukemia in which malignant megakaryoblasts proliferate abnormally and injure various tissues. Megakaryoblasts are the most immature precursor cells in a platelet-forming lineage; they mature to promegakaryocytes and, ultimately, megakaryocytes which cells shed membrane-enclosed particles, i.e. platelets, into the circulation. Platelets are critical for the normal clotting of blood. While malignant megakaryoblasts usually are the predominant proliferating and tissue-damaging cells, their similarly malignant descendants, promegakaryocytes and megakaryocytes, are variable contributors to the malignancy.
MonoMAC syndrome is a rare autosomal dominant syndrome associated with: monocytopenia, B and NK cell lymphopenia; mycobacterial, viral, fungal, and bacterial opportunistic infections; and virus infection-induced cancers. The disorder often progresses to the development of myelodysplasia, myeloid leukemias, and other types of cancer. MonoMAC is a life-threatening and precancerous disorder.
Clonal hypereosinophilia, also termed primary hypereosinophilia or clonal eosinophilia, is a grouping of hematological disorders all of which are characterized by the development and growth of a pre-malignant or malignant population of eosinophils, a type of white blood cell that occupies the bone marrow, blood, and other tissues. This population consists of a clone of eosinophils, i.e. a group of genetically identical eosinophils derived from a sufficiently mutated ancestor cell.

The Emberger syndrome is a rare, autosomal dominant, genetic disorder caused by familial or sporadic inactivating mutations in one of the two parental GATA2 genes. The mutation results in a haploinsufficiency in the levels of the gene's product, the GATA2 transcription factor. This transcription factor is critical for the embryonic development, maintenance, and functionality of blood-forming, lympathic-forming, and other tissues. The syndrome includes as its primary symptoms: serious abnormalities of the blood such as the myelodysplastic syndrome and acute myeloid leukemia; lymphedema of the lower limbs, and sensorineural hearing loss. However, the anomalies caused by GATA2 mutations are highly variable with some individuals showing little or no such symptoms even in old age while others exhibit non-malignant types of hematological anomalies; lymphedema in areas besides the lower limbs, little or no hearing loss; or anomalies in other tissues. The syndrome may present with relatively benign signs and/or symptoms and then progress rapidly or slowly to the myelodysplastic syndrome and/or acute myeloid leukemia. Alternatively, it may present with one of the latter two life-threatening disorders.
Transient myeloproliferative disease (TMD) occurs in a significant percentage of individuals born with the congenital genetic disorder, Down syndrome. It may occur in individuals who are not diagnosed with the syndrome but have some hematological cells containing genetic abnormalities that are similar to those found in Down syndrome. TMD usually develops in utero, is diagnosed prenatally or within ~3 months of birth, and thereafter resolves rapidly and spontaneously. However, during the prenatal-to-postnatal period, the disease may cause irreparable damage to various organs and in ~20% of individuals death. Moreover, ~10% of individuals diagnosed with TMD develop acute megakaryoblastic leukemia at some time during the 5 years following its resolution. TMD is a life-threatening, precancerous condition in fetuses as well as infants in their first few months of life.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Crispino JD, Horwitz MS (April 2017). "GATA factor mutations in hematologic disease". Blood. 129 (15): 2103–2110. doi:10.1182/blood-2016-09-687889. PMC 5391620 . PMID 28179280.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 Hirabayashi S, Wlodarski MW, Kozyra E, Niemeyer CM (August 2017). "Heterogeneity of GATA2-related myeloid neoplasms". International Journal of Hematology. 106 (2): 175–182. doi: 10.1007/s12185-017-2285-2 . PMID 28643018.
- ↑ Bannon SA, DiNardo CD (May 2016). "Hereditary Predispositions to Myelodysplastic Syndrome". International Journal of Molecular Sciences. 17 (6): 838. doi: 10.3390/ijms17060838 . PMC 4926372 . PMID 27248996.
- 1 2 West AH, Godley LA, Churpek JE (March 2014). "Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations". Annals of the New York Academy of Sciences. 1310 (1): 111–8. Bibcode:2014NYASA1310..111W. doi:10.1111/nyas.12346. PMC 3961519 . PMID 24467820.
- 1 2 3 Camargo JF, Lobo SA, Hsu AP, Zerbe CS, Wormser GP, Holland SM (September 2013). "MonoMAC syndrome in a patient with a GATA2 mutation: case report and review of the literature". Clinical Infectious Diseases. 57 (5): 697–9. doi:10.1093/cid/cit368. PMC 3739466 . PMID 23728141.
- 1 2 Johnson JA, Yu SS, Elist M, Arkfeld D, Panush RS (September 2015). "Rheumatologic manifestations of the "MonoMAC" syndrome. a systematic review". Clinical Rheumatology. 34 (9): 1643–5. doi:10.1007/s10067-015-2905-2. PMID 25739845. S2CID 29935351.
- 1 2 3 4 5 Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, Dafou D, Kilo T, Smithson S, Lunt P, Murday VA, Hodgson S, Keenan R, Pilz DT, Martinez-Corral I, Makinen T, Mortimer PS, Jeffery S, Trembath RC, Mansour S (September 2011). "Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome)" (PDF). Nature Genetics. 43 (10): 929–31. doi:10.1038/ng.923. PMID 21892158. S2CID 23449974.
- 1 2 3 Mansour S, Connell F, Steward C, Ostergaard P, Brice G, Smithson S, Lunt P, Jeffery S, Dokal I, Vulliamy T, Gibson B, Hodgson S, Cottrell S, Kiely L, Tinworth L, Kalidas K, Mufti G, Cornish J, Keenan R, Mortimer P, Murday V (September 2010). "Emberger syndrome-primary lymphedema with myelodysplasia: report of seven new cases". American Journal of Medical Genetics. Part A. 152A (9): 2287–96. doi:10.1002/ajmg.a.33445. PMID 20803646. S2CID 205312771.
- 1 2 3 4 5 Mir MA, Kochuparambil ST, Abraham RS, Rodriguez V, Howard M, Hsu AP, Jackson AE, Holland SM, Patnaik MM (April 2015). "Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations". Cancer Medicine. 4 (4): 490–9. doi:10.1002/cam4.384. PMC 4402062 . PMID 25619630.
- 1 2 3 Locatelli F, Strahm B (March 2018). "How I treat myelodysplastic syndromes of childhood". Blood. 131 (13): 1406–1414. doi: 10.1182/blood-2017-09-765214 . hdl: 11573/1480317 . PMID 29438960.
- 1 2 3 4 5 Hasle H (December 2016). "Myelodysplastic and myeloproliferative disorders of childhood". Hematology. American Society of Hematology. Education Program. 2016 (1): 598–604. doi:10.1182/asheducation-2016.1.598. PMC 6142519 . PMID 27913534.
- ↑ Churpek JE (December 2017). "Familial myelodysplastic syndrome/acute myeloid leukemia". Best Practice & Research. Clinical Haematology. 30 (4): 287–289. doi:10.1016/j.beha.2017.10.002. PMC 5774636 . PMID 29156196.
- 1 2 Donadieu J, Beaupain B, Fenneteau O, Bellanné-Chantelot C (November 2017). "Congenital neutropenia in the era of genomics: classification, diagnosis, and natural history". British Journal of Haematology. 179 (4): 557–574. doi: 10.1111/bjh.14887 . PMID 28875503. S2CID 1477026.
- ↑ Fisher KE, Hsu AP, Williams CL, Sayeed H, Merritt BY, Elghetany MT, Holland SM, Bertuch AA, Gramatges MM (February 2017). "Somatic mutations in children with GATA2-associated myelodysplastic syndrome who lack other features of GATA2 deficiency". Blood Advances. 1 (7): 443–448. doi:10.1182/bloodadvances.2016002311. PMC 5738979 . PMID 29296959.
- 1 2 3 4 5 6 7 8 9 10 Hsu AP, McReynolds LJ, Holland SM (February 2015). "GATA2 deficiency". Current Opinion in Allergy and Clinical Immunology. 15 (1): 104–9. doi:10.1097/ACI.0000000000000126. PMC 4342850 . PMID 25397911.
- 1 2 3 4 5 6 7 Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, Arthur DC, Gu W, Gould CM, Brewer CC, Cowen EW, Freeman AF, Olivier KN, Uzel G, Zelazny AM, Daub JR, Spalding CD, Claypool RJ, Giri NK, Alter BP, Mace EM, Orange JS, Cuellar-Rodriguez J, Hickstein DD, Holland SM (February 2014). "GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity". Blood. 123 (6): 809–21. doi:10.1182/blood-2013-07-515528. PMC 3916876 . PMID 24227816.
- ↑ Brambila-Tapia AJ, García-Ortiz JE, Brouillard P, Nguyen HL, Vikkula M, Ríos-González BE, Sandoval-Muñiz RJ, Sandoval-Talamantes AK, Bobadilla-Morales L, Corona-Rivera JR, Arnaud-Lopez L (September 2017). "GATA2 null mutation associated with incomplete penetrance in a family with Emberger syndrome". Hematology (Amsterdam, Netherlands). 22 (8): 467–471. doi: 10.1080/10245332.2017.1294551 . PMID 28271814.
- 1 2 3 4 Bigley V, Cytlak U, Collin M (February 2018). "Human dendritic cell immunodeficiencies". Seminars in Cell & Developmental Biology. 86: 50–61. doi:10.1016/j.semcdb.2018.02.020. PMID 29452225. S2CID 3557136.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Wlodarski MW, Collin M, Horwitz MS (April 2017). "GATA2 deficiency and related myeloid neoplasms". Seminars in Hematology. 54 (2): 81–86. doi:10.1053/j.seminhematol.2017.05.002. PMC 5650112 . PMID 28637621.
- 1 2 3 4 5 6 7 Katsumura KR, Bresnick EH (April 2017). "The GATA factor revolution in hematology". Blood. 129 (15): 2092–2102. doi:10.1182/blood-2016-09-687871. PMC 5391619 . PMID 28179282.
- 1 2 Shimizu R, Yamamoto M (August 2016). "GATA-related hematologic disorders". Experimental Hematology. 44 (8): 696–705. doi: 10.1016/j.exphem.2016.05.010 . PMID 27235756.
- 1 2 Chlon TM, Crispino JD (November 2012). "Combinatorial regulation of tissue specification by GATA and FOG factors". Development. 139 (21): 3905–16. doi:10.1242/dev.080440. PMC 3472596 . PMID 23048181.
- ↑ "GATA2 GATA binding protein 2 [Homo sapiens (human)] - Gene - NCBI".
- ↑ Katoh M (July 2013). "Functional and cancer genomics of ASXL family members". British Journal of Cancer. 109 (2): 299–306. doi: 10.1038/bjc.2013.281 . PMC 3721406 . PMID 23736028.
- ↑ Wang L, Du F, Zhang HM, Wang HX (July 2015). "Evaluation of a father and son with atypical chronic myeloid leukemia with SETBP1 mutations and a review of the literature". Brazilian Journal of Medical and Biological Research. 48 (7): 583–7. doi:10.1590/1414-431X20154557. PMC 4512095 . PMID 26017341.
- ↑ Viny AD, Levine RL (March 2018). "Cohesin mutations in myeloid malignancies made simple". Current Opinion in Hematology. 25 (2): 61–66. doi:10.1097/MOH.0000000000000405. PMC 6601335 . PMID 29278534.
- ↑ Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, Jardine L, Pagan S, Dimmick I, Chua I, Wallis J, Lordan J, Morgan C, Kumararatne DS, Doffinger R, van der Burg M, van Dongen J, Cant A, Dick JE, Hambleton S, Collin M (February 2011). "The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency". The Journal of Experimental Medicine. 208 (2): 227–34. doi:10.1084/jem.20101459. PMC 3039861 . PMID 21242295.
- ↑ Moriguchi T, Yamamoto M (November 2014). "A regulatory network governing Gata1 and Gata2 gene transcription orchestrates erythroid lineage differentiation". International Journal of Hematology. 100 (5): 417–24. doi: 10.1007/s12185-014-1568-0 . PMID 24638828.
- 1 2 3 4 Babushok DV, Bessler M (March 2015). "Genetic predisposition syndromes: when should they be considered in the work-up of MDS?". Best Practice & Research. Clinical Haematology. 28 (1): 55–68. doi:10.1016/j.beha.2014.11.004. PMC 4323616 . PMID 25659730.
- 1 2 3 Rastogi N, Abraham RS, Chadha R, Thakkar D, Kohli S, Nivargi S, Prakash Yadav S (November 2017). "Successful Nonmyeloablative Allogeneic Stem Cell Transplant in a Child With Emberger Syndrome and GATA2 Mutation". Journal of Pediatric Hematology/Oncology. 40 (6): e383–e388. doi:10.1097/MPH.0000000000000995. PMID 29189513. S2CID 19080505.
- ↑ Porter CC (December 2016). "Germ line mutations associated with leukemias". Hematology. American Society of Hematology. Education Program. 2016 (1): 302–308. doi:10.1182/asheducation-2016.1.302. PMC 6142470 . PMID 27913495.
- ↑ m. d, Dennis Hickstein (4 March 2020). "Pilot and Feasibility Study of Reduced-Intensity Hematopoietic Stem Cell Transplant for Patients with GATA2 Mutations".
- ↑ "Allogeneic Hematopoietic Stem Cell Transplant for Patients with Mutations in GATA2 or the MonoMAC Syndrome". 20 May 2021.
- ↑ Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, Lakey JH, Rahman T, Wang XN, McGovern N, Pagan S, Cookson S, McDonald D, Chua I, Wallis J, Cant A, Wright M, Keavney B, Chinnery PF, Loughlin J, Hambleton S, Santibanez-Koref M, Collin M (September 2011). "Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency". Blood. 118 (10): 2656–8. doi:10.1182/blood-2011-06-360313. PMC 5137783 . PMID 21765025.
- ↑ Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, Babic M, Lin M, Carmagnac A, Lee YK, Kok CH, Gagliardi L, Friend KL, Ekert PG, Butcher CM, Brown AL, Lewis ID, To LB, Timms AE, Storek J, Moore S, Altree M, Escher R, Bardy PG, Suthers GK, D'Andrea RJ, Horwitz MS, Scott HS (September 2011). "Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia". Nature Genetics. 43 (10): 1012–7. doi:10.1038/ng.913. PMC 3184204 . PMID 21892162.