In organic chemistry, a carboxylic acid is an organic acid that contains a carboxyl group attached to an R-group. The general formula of a carboxylic acid is often written as R−COOH or R−CO2H, sometimes as R−C(O)OH with R referring to the alkyl, alkenyl, aryl, or other group. Carboxylic acids occur widely. Important examples include the amino acids and fatty acids. Deprotonation of a carboxylic acid gives a carboxylate anion.
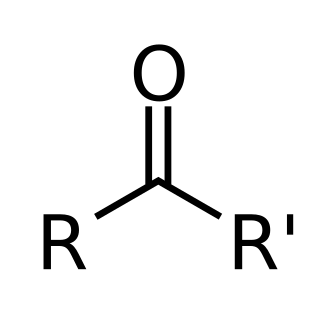
In organic chemistry, a ketone is an organic compound with the structure R−C(=O)−R', where R and R' can be a variety of carbon-containing substituents. Ketones contain a carbonyl group −C(=O)−. The simplest ketone is acetone, with the formula (CH3)2CO. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids, and the solvent acetone.
The Friedel–Crafts reactions are a set of reactions developed by Charles Friedel and James Crafts in 1877 to attach substituents to an aromatic ring. Friedel–Crafts reactions are of two main types: alkylation reactions and acylation reactions. Both proceed by electrophilic aromatic substitution.

The aldol reaction is a reaction in organic chemistry that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. Its simplest form might involve the nucleophilic addition of an enolized ketone to another:

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.

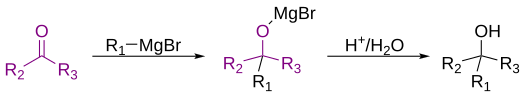
Francois Auguste Victor Grignard was a French chemist who won the Nobel Prize for his discovery of the eponymously named Grignard reagent and Grignard reaction, both of which are important in the formation of carbon–carbon bonds.

In organic chemistry, enolates are organic anions derived from the deprotonation of carbonyl compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds.
Nucleophilic acyl substitution describes a class of substitution reactions involving nucleophiles and acyl compounds. In this type of reaction, a nucleophile – such as an alcohol, amine, or enolate – displaces the leaving group of an acyl derivative – such as an acid halide, anhydride, or ester. The resulting product is a carbonyl-containing compound in which the nucleophile has taken the place of the leaving group present in the original acyl derivative. Because acyl derivatives react with a wide variety of nucleophiles, and because the product can depend on the particular type of acyl derivative and nucleophile involved, nucleophilic acyl substitution reactions can be used to synthesize a variety of different products.
The Weinreb ketone synthesis or Weinreb–Nahm ketone synthesis is a chemical reaction used in organic chemistry to make carbon–carbon bonds. It was discovered in 1981 by Steven M. Weinreb and Steven Nahm as a method to synthesize ketones. The original reaction involved two subsequent nucleophilic acyl substitutions: the conversion of an acid chloride with N,O-Dimethylhydroxylamine, to form a Weinreb–Nahm amide, and subsequent treatment of this species with an organometallic reagent such as a Grignard reagent or organolithium reagent. Nahm and Weinreb also reported the synthesis of aldehydes by reduction of the amide with an excess of lithium aluminum hydride.
The Blaise ketone synthesis is the chemical reaction of acid chlorides with organozinc compounds to give ketones.
The Sakurai reaction is the chemical reaction of carbon electrophiles with allyltrimethylsilane catalyzed by strong Lewis acids. The reaction achieves results similar to the addition of an allyl Grignard reagent to the carbonyl.

Grignard reagents or Grignard compounds are chemical compounds with the general formula R−Mg−X, where X is a halogen and R is an organic group, normally an alkyl or aryl. Two typical examples are methylmagnesium chloride Cl−Mg−CH3 and phenylmagnesium bromide (C6H5)−Mg−Br. They are a subclass of the organomagnesium compounds.
In organosilicon chemistry, silyl enol ethers are a class of organic compounds that share the common functional group R3Si−O−CR=CR2, composed of an enolate bonded to a silane through its oxygen end and an ethene group as its carbon end. They are important intermediates in organic synthesis.

Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.

The Stork enamine alkylation involves the addition of an enamine to a Michael acceptor or another electrophilic alkylation reagent to give an alkylated iminium product, which is hydrolyzed by dilute aqueous acid to give the alkylated ketone or aldehyde. Since enamines are generally produced from ketones or aldehydes, this overall process constitutes a selective monoalkylation of a ketone or aldehyde, a process that may be difficult to achieve directly.
Organomanganese chemistry is the chemistry of organometallic compounds containing a carbon to manganese chemical bond. In a 2009 review, Cahiez et al. argued that as manganese is cheap and benign, organomanganese compounds have potential as chemical reagents, although currently they are not widely used as such despite extensive research.
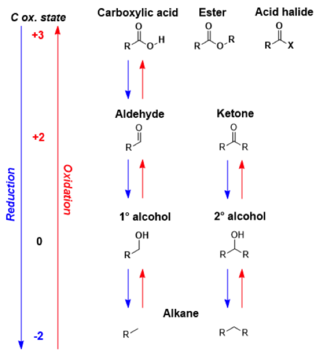
In organic chemistry, carbonyl reduction is the conversion of any carbonyl group, usually to an alcohol. It is a common transformation that is practiced in many ways. Ketones, aldehydes, carboxylic acids, esters, amides, and acid halides - some of the most pervasive functional groups, -comprise carbonyl compounds. Carboxylic acids, esters, and acid halides can be reduced to either aldehydes or a step further to primary alcohols, depending on the strength of the reducing agent. Aldehydes and ketones can be reduced respectively to primary and secondary alcohols. In deoxygenation, the alcohol group can be further reduced and removed altogether by replacement with H.

Reductions with samarium(II) iodide involve the conversion of various classes of organic compounds into reduced products through the action of samarium(II) iodide, a mild one-electron reducing agent.
Reactions of organocopper reagents involve species containing copper-carbon bonds acting as nucleophiles in the presence of organic electrophiles. Organocopper reagents are now commonly used in organic synthesis as mild, selective nucleophiles for substitution and conjugate addition reactions.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.: