
Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Spinal muscular atrophies (SMAs) are a genetically and clinically heterogeneous group of rare debilitating disorders characterised by the degeneration of lower motor neurons and subsequent atrophy (wasting) of various muscle groups in the body. While some SMAs lead to early infant death, other diseases of this group permit normal adult life with only mild weakness.
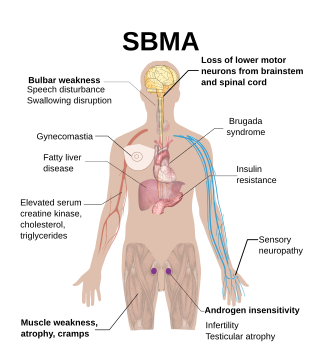
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.

A neuromuscular disease is any disease affecting the peripheral nervous system (PNS), the neuromuscular junctions, or skeletal muscles, all of which are components of the motor unit. Damage to any of these structures can cause muscle atrophy and weakness. Issues with sensation can also occur.
Spinal muscular atrophy (SMA) is a rare neuromuscular disorder that results in the loss of motor neurons and progressive muscle wasting. It is usually diagnosed in infancy or early childhood and if left untreated it is the most common genetic cause of infant death. It may also appear later in life and then have a milder course of the disease. The common feature is progressive weakness of voluntary muscles, with arm, leg and respiratory muscles being affected first. Associated problems may include poor head control, difficulties swallowing, scoliosis, and joint contractures.

Fazio–Londe disease (FLD), also called progressive bulbar palsy of childhood, is a very rare inherited motor neuron disease of children and young adults and is characterized by progressive paralysis of muscles innervated by cranial nerves. FLD, along with Brown–Vialetto–Van Laere syndrome (BVVL), are the two forms of infantile progressive bulbar palsy, a type of progressive bulbar palsy in children.

Wilhelm Heinrich Erb was a German neurologist. He was born in Winnweiler, and died in Heidelberg.

Hereditary motor and sensory neuropathies (HMSN) is a name sometimes given to a group of different neuropathies which are all characterized by their impact upon both afferent and efferent neural communication. HMSN are characterised by atypical neural development and degradation of neural tissue. The two common forms of HMSN are either hypertrophic demyelinated nerves or complete atrophy of neural tissue. Hypertrophic condition causes neural stiffness and a demyelination of nerves in the peripheral nervous system, and atrophy causes the breakdown of axons and neural cell bodies. In these disorders, a patient experiences progressive muscle atrophy and sensory neuropathy of the extremities.

X-linked spinal muscular atrophy type 2, also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.
Distal spinal muscular atrophy type 1 (DSMA1), also known as spinal muscular atrophy with respiratory distress type 1 (SMARD1), is a rare neuromuscular disorder involving death of motor neurons in the spinal cord which leads to a generalised progressive atrophy of body muscles.
Distal hereditary motor neuronopathies, sometimes also called distal hereditary motor neuropathies, are a genetically and clinically heterogeneous group of motor neuron diseases that result from genetic mutations in various genes and are characterized by degeneration and loss of motor neuron cells in the anterior horn of the spinal cord and subsequent muscle atrophy.
Congenital distal spinal muscular atrophy (cDSMA), also known as distal hereditary motor neuropathytype VIII (dHMN8), is a hereditary medical condition characterized by muscle wasting (atrophy), particularly of distal muscles in legs and hands, and by early-onset contractures of the hip, knee, and ankle. Affected individuals often have shorter lower limbs relative to the trunk and upper limbs. The condition is a result of a loss of anterior horn cells localized to lumbar and cervical regions of the spinal cord early in infancy, which in turn is caused by a mutation of the TRPV4 gene. The disorder is inherited in an autosomal dominant manner. Arm muscle and function, as well as cardiac and respiratory functions are typically well preserved.
Jokela is a town in Tuusula, Finland.
Distal spinal muscular atrophy type 2 (DSMA2), also known as Jerash type distal hereditary motor neuropathy (HMNJ), is a very rare childhood-onset genetic disorder characterised by progressive muscle wasting affecting lower and subsequently upper limbs. The disorder has been described in Arab inhabitants of Jerash region in Jordan as well as in a Chinese family.
Coiled-coil-helix-coiled-coil-helix domain-containing protein 10, mitochondrial, also known as Protein N27C7-4 is a protein that in humans is encoded by the CHCHD10 gene.

Charlotte Jane Sumner is an American neurologist. She is a professor in the Departments of Neurology and Neuroscience at Johns Hopkins School of Medicine. Dr. Sumner cares for patients with genetically mediated neuromuscular diseases and directs a laboratory focused on developing treatments for these diseases. She co-directs the Johns Hopkins Muscular Dystrophy Association Care Center, the Spinal Muscular Atrophy (SMA), and the Charcot-Marie-Tooth (CMT) clinics, which deliver multidisciplinary clinical care, engage in international natural history studies, and provide cutting edge therapeutics.

Spinal muscular atrophy with lower extremity predominance 2A (SMALED2A) is a rare neuromuscular disorder characterised by muscle weakness predominantly in legs. The disorder is usually diagnosed shortly after birth; affected children have a delayed motor development, waddling gait, difficulties walking, sometimes develop spasticity. Sensation, swallowing and cognitive development are not affected. The disorder is slowly progressive throughout the lifetime.
Spinal muscular atrophy with lower extremity predominance 2B (SMALED2B) is a rare neuromuscular disorder characterised by generalised muscle weakness.