Related Research Articles

Frontotemporal dementia (FTD), or frontotemporal degeneration disease, or frontotemporal neurocognitive disorder, encompasses several types of dementia involving the progressive degeneration of frontal and temporal lobes. FTDs broadly present as behavioral or language disorders with gradual onsets. Common signs and symptoms include significant changes in social and personal behavior, apathy, blunting of emotions, and deficits in both expressive and receptive language. Currently, there is no cure for FTD, but there are treatments that help alleviate symptoms.
Degenerative disease is the result of a continuous process based on degenerative cell changes, affecting tissues or organs, which will increasingly deteriorate over time.

Frontotemporal lobar degeneration (FTLD) is a pathological process that occurs in frontotemporal dementia. It is characterized by atrophy in the frontal lobe and temporal lobe of the brain, with sparing of the parietal and occipital lobes.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.
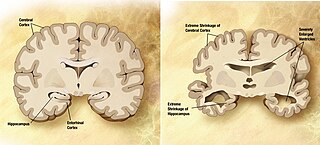
A neurodegenerative disease is caused by the progressive loss of structure or function of neurons, in the process known as neurodegeneration. Such neuronal damage may ultimately involve cell death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

TIA1 or Tia1 cytotoxic granule-associated rna binding protein is a 3'UTR mRNA binding protein that can bind the 5'TOP sequence of 5'TOP mRNAs. It is associated with programmed cell death (apoptosis) and regulates alternative splicing of the gene encoding the Fas receptor, an apoptosis-promoting protein. Under stress conditions, TIA1 localizes to cellular RNA-protein conglomerations called stress granules. It is encoded by the TIA1 gene.

Superoxide dismutase [Cu-Zn] also known as superoxide dismutase 1 or hSod1 is an enzyme that in humans is encoded by the SOD1 gene, located on chromosome 21. SOD1 is one of three human superoxide dismutases. It is implicated in apoptosis, familial amyotrophic lateral sclerosis and Parkinson's disease.

Dynamin-like 120 kDa protein, mitochondrial is a protein that in humans is encoded by the OPA1 gene. This protein regulates mitochondrial fusion and cristae structure in the inner mitochondrial membrane (IMM) and contributes to ATP synthesis and apoptosis, and small, round mitochondria. Mutations in this gene have been implicated in dominant optic atrophy (DOA), leading to loss in vision, hearing, muscle contraction, and related dysfunctions.

ATP synthase subunit delta, mitochondrial, also known as ATP synthase F1 subunit delta or F-ATPase delta subunit is an enzyme that in humans is encoded by the ATP5F1D gene. This gene encodes a subunit of mitochondrial ATP synthase. Mitochondrial ATP synthase catalyzes ATP synthesis, utilizing an electrochemical gradient of protons across the inner membrane during oxidative phosphorylation.

Transmembrane protein 106B is a protein that is encoded by the TMEM106B gene. It is found primarily within neurons and oligodendrocytes in the central nervous system with its subcellular location being in lysosomal membranes. TMEM106B helps facilitate important functions for maintaining a healthy lysosome, and therefore certain mutations and polymorphisms can lead to issues with proper lysosomal function. Lysosomes are in charge of clearing out mis-folded proteins and other debris, and thus, play an important role in neurodegenerative diseases that are driven by the accumulation of various mis-folded proteins and aggregates. Due to its impact on lysosomal function, TMEM106B has been investigated and found to be associated to multiple neurodegenerative diseases.

Amyotrophic lateral sclerosis (ALS), also known as motor neurone disease (MND) or Lou Gehrig's disease, is a rare and terminal neurodegenerative disease that results in the progressive loss of motor neurons that control voluntary muscles. ALS is the most common form of the motor neuron diseases. Early symptoms of ALS include stiff muscles, muscle twitches, gradual increasing weakness, and muscle wasting. Limb-onset ALS begins with weakness in the arms or legs, while bulbar-onset ALS begins with difficulty in speaking or swallowing. Around half of people with ALS develop at least mild difficulties with thinking and behavior, and about 15% develop frontotemporal dementia. Motor neuron loss continues until the abilities to eat, speak, move, or, lastly, breathe are lost.
Mitochondrially encoded tRNA isoleucine also known as MT-TI is a transfer RNA which in humans is encoded by the mitochondrial MT-TI gene.

C9orf72 is a protein which in humans is encoded by the gene C9orf72.
Multisystem proteinopathy (MSP) is a dominantly inherited, pleiotropic, degenerative disorder of humans that can affect muscle, bone, and/or the central nervous system. MSP can manifest clinically as classical amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), inclusion body myopathy (IBM), Paget's disease of bone (PDB), or as a combination of these disorders. Historically, several different names have been used to describe MSP, most commonly "inclusion body myopathy with early-onset Paget disease and frontotemporal dementia (IBMPFD)" or "inclusion body myopathy with frontotemporal dementia, Paget's disease of bone, and amyotrophic lateral sclerosis (IBMPFD/ALS)." However, IBMPFD and IBMPFD/ALS are now considered outdated classifications and are more properly referred to as MSP, as the disease is clinically heterogeneous and its phenotypic spectrum extends beyond IBM, PDB, FTD, and ALS to include motor neuron disease, Parkinson's disease features, and ataxia features. Although MSP is rare, growing interest in this syndrome derives from the molecular insights the condition provides into the etiological relationship between common age-related degenerative diseases of muscle, bone, and brain.
Jokela type spinal muscular atrophy (SMAJ), also known as late-onset spinal motor neuronopathy (LOSMoN), is an ultra-rare neuromuscular disorder characterized by muscle twitches and cramps. The symptoms appear in adulthood and gradually progress. The disease is caused by a mutation in the CHCHD10 gene and is inherited in an autosomal dominant pattern. It was first described by the Finnish neurologist Manu Jokela in 2011.
There are more than 25 genes known to be associated with amyotrophic lateral sclerosis (ALS) as of June 2018, which collectively account for about 70% of cases of familial ALS (fALS) and 10% of cases of sporadic ALS (sALS). About 5–10% of cases of ALS are directly inherited. Overall, first-degree relatives of an individual with ALS have a 1% risk of developing ALS. ALS has an oligogenic mode of inheritance, meaning that mutations in two or more genes are required to cause disease.

Cytochrome c oxidase assembly factor 7 (putative) (COA7), also known as Beta-lactamase hap-like protein, Respiratory chain assembly factor 1 (RESA1), Sel1 repeat-containing protein 1 (SELRC1), or C1orf163 is a protein that in humans is encoded by the COA7 gene. The protein encoded by COA7 is an assembly factor important for the mitochondrial respiratory chain. Mutations in COA7 have been associated with cytochrome c oxidase deficiency resulting in spinocerebellar ataxia with axonal neuropathy type 3 and mitochondrial myopathy.

Solute carrier family 25 member 46 is a protein that in humans is encoded by the SLC25A46 gene. This protein is a member of the SLC25 mitochondrial solute carrier family. It is a transmembrane protein located in the mitochondrial outer membrane involved in lipid transfer from the endoplasmic reticulum (ER) to mitochondria. Mutations in this gene result in neuropathy and optic atrophy.
References
- 1 2 3 4 "Entrez Gene: coiled-coil-helix-coiled-coil-helix domain containing 10" . Retrieved 2018-08-14.
 This article incorporates text from this source, which is in the public domain .
This article incorporates text from this source, which is in the public domain . - 1 2 3 4 5 6 7 "CHCHD10 - Coiled-coil-helix-coiled-coil-helix domain-containing protein 10, mitochondrial precursor - Homo sapiens (Human) - CHCHD10 gene & protein" . Retrieved 2018-08-07.
 This article incorporates text available under the CC BY 4.0 license.
This article incorporates text available under the CC BY 4.0 license. - 1 2 3 4 5 6 7 "UniProt: the universal protein knowledgebase". Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571 . PMID 27899622.
- ↑ Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem D, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss J, Duan H, Uhlen M, Yates JR, Apweiler R, Ge J, Hermjakob H, Ping P (Oct 2013). "Integration of cardiac proteome biology and medicine by a specialized knowledgebase". Circulation Research. 113 (9): 1043–53. doi:10.1161/CIRCRESAHA.113.301151. PMC 4076475 . PMID 23965338.
- ↑ "Coiled-coil-helix-coiled-coil-helix domain-containing protein 10, mitochondrial". Cardiac Organellar Protein Atlas Knowledgebase (COPaKB).[ permanent dead link ]
- ↑ Bannwarth, S; Ait-El-Mkadem, S; Chaussenot, A; Genin, EC; Lacas-Gervais, S; Fragaki, K; Berg-Alonso, L; Kageyama, Y; Serre, V; Moore, DG; Verschueren, A; Rouzier, C; Le Ber, I; Augé, G; Cochaud, C; Lespinasse, F; N'Guyen, K; de Septenville, A; Brice, A; Yu-Wai-Man, P; Sesaki, H; Pouget, J; Paquis-Flucklinger, V (August 2014). "A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement". Brain: A Journal of Neurology. 137 (Pt 8): 2329–45. doi:10.1093/brain/awu138. PMC 4107737 . PMID 24934289.
- ↑ Ajroud-Driss, Senda; Fecto, Faisal (6 September 2014). "Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy". Neurogenetics. 16 (1): 1–9. doi:10.1007/s10048-014-0421-1. PMC 4796476 . PMID 25193783.
Frontotemporal dementia and/or amyotrophic lateral sclerosis 2 (FTDALS2)
- 1 2 Bannwarth, Sylvie; Ait-El-Mkadem, Samira (16 June 2014). "A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement". Brain. 137 (8): 2329–2345. doi:10.1093/brain/awu138. PMC 4107737 . PMID 24934289.
- ↑ Mick DU, Dennerlein S, Wiese H, Reinhold R, Pacheu-Grau D, Lorenzi I, Sasarman F, Weraarpachai W, Shoubridge EA, Warscheid B, Rehling P (December 2012). "MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation". Cell. 151 (7): 1528–41. doi: 10.1016/j.cell.2012.11.053 . hdl: 11858/00-001M-0000-000E-DDDF-4 . PMID 23260140.
This article incorporates text from the United States National Library of Medicine (), which is in the public domain.