Related Research Articles
Quantum chemistry, also called molecular quantum mechanics, is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
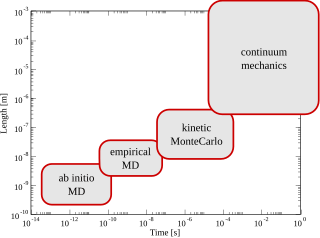
Multiscale modeling or multiscale mathematics is the field of solving problems that have important features at multiple scales of time and/or space. Important problems include multiscale modeling of fluids, solids, polymers, proteins, nucleic acids as well as various physical and chemical phenomena.
The Molecular Modelling Toolkit (MMTK) is an open-source software package written in Python, which performs common tasks in molecular modelling.
The Molecular Modeling Toolkit is a library that implements common molecular simulation techniques, with an emphasis on biomolecular simulations. It uses modern software engineering techniques in order to overcome limitations associated with the large monolithic simulation programs that are commonly used for biomolecules. Its principal advantages are (1) easy extension and combination with other libraries due to modular library design, (2) a single high-level general-purpose programming language (Python) is used for library implementation as well as for application scripts, (3) use of documented and machine-independent formats for all data files, and (4) interfaces to other simulation and visualization programs.
The kinetic Monte Carlo (KMC) method is a Monte Carlo method computer simulation intended to simulate the time evolution of some processes occurring in nature. Typically these are processes that occur with known transition rates among states. It is important to understand that these rates are inputs to the KMC algorithm, the method itself cannot predict them.
Tinker, previously stylized as TINKER, is a suite of computer software applications for molecular dynamics simulation. The codes provide a complete and general set of tools for molecular mechanics and molecular dynamics, with some special features for biomolecules. The core of the software is a modular set of callable routines which allow manipulating coordinates and evaluating potential energy and derivatives via straightforward means.
MBN may refer to:
This is a list of computer programs that are predominantly used for molecular mechanics calculations.
Materials Studio is software for simulating and modeling materials. It is developed and distributed by BIOVIA, a firm specializing in research software for computational chemistry, bioinformatics, cheminformatics, molecular dynamics simulation, and quantum mechanics.
Nanomechanics is a branch of nanoscience studying fundamental mechanical properties of physical systems at the nanometer scale. Nanomechanics has emerged on the crossroads of biophysics, classical mechanics, solid-state physics, statistical mechanics, materials science, and quantum chemistry. As an area of nanoscience, nanomechanics provides a scientific foundation of nanotechnology.
This is a list of notable computer programs that are used for nucleic acids simulations.

Ascalaph Designer is a computer program for general purpose molecular modelling for molecular design and simulations. It provides a graphical environment for the common programs of quantum and classical molecular modelling ORCA, NWChem, Firefly, CP2K and MDynaMix . The molecular mechanics calculations cover model building, energy optimizations and molecular dynamics. Firefly covers a wide range of quantum chemistry methods. Ascalaph Designer is free and open-source software, released under the GNU General Public License, version 2 (GPLv2).
MacroModel is a computer program for molecular modelling of organic compounds and biopolymers. It features various chemistry force fields, plus energy minimizing algorithms, to predict geometry and relative conformational energies of molecules. MacroModel is maintained by Schrödinger, LLC.
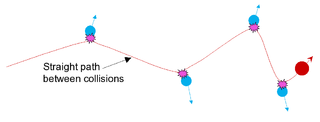
In condensed-matter physics, the binary collision approximation (BCA) is a heuristic used to more efficiently simulate the penetration depth and defect production by energetic ions in solids. In the method, the ion is approximated to travel through a material by experiencing a sequence of independent binary collisions with sample atoms (nuclei). Between the collisions, the ion is assumed to travel in a straight path, experiencing electronic stopping power, but losing no energy in collisions with nuclei.
Multi-state modeling of biomolecules refers to a series of techniques used to represent and compute the behaviour of biological molecules or complexes that can adopt a large number of possible functional states.
Polymer crystals have different properties than simple atomic crystals. They possess high density and long range order. They do not possess isotropy, and therefore are anisotropic in nature, which means they show anisotropy and limited conformation space. However, just as atomic crystals have lattices, polymer crystals also exhibit a periodic structure called a lattice, which describes the repetition of the unit cells in the space. The simulation of polymer crystals is complex and not taken from only one state but from solid-state and fluid-state physics as well. Polymer crystals have unit cells that consist of tens of atoms, while the molecules themselves comprise 104 To 106 atoms.
Computational materials science and engineering uses modeling, simulation, theory, and informatics to understand materials. The main goals include discovering new materials, determining material behavior and mechanisms, explaining experiments, and exploring materials theories. It is analogous to computational chemistry and computational biology as an increasingly important subfield of materials science.
In mathematics and physics, surface growth refers to models used in the dynamical study of the growth of a surface, usually by means of a stochastic differential equation of a field.
References
- ↑ "About MBN Explorer". mbnresearch.com. Retrieved 31 August 2017.
- ↑ "MBN Explorer: A decade development now available for the community". Virtual Institute of Nano Films. Retrieved 31 August 2017.
- ↑ I.A. Solov'yov; A.V. Yakubovich; P.V. Nikolaev; I. Volkovets; A.V. Solov'yov (2012). "MesoBioNano Explorer - A universal program for multiscale computer simulations of complex molecular structure and dynamics". J. Comput. Chem. 33 (30): 2412–2439. doi:10.1002/jcc.23086. PMID 22965786. S2CID 22553279.
- ↑ M. Panshenskov; I.A. Solov'yov; A.V. Solov'yov (2014). "Efficient 3D kinetic monte carlo method for modeling of molecular structure and dynamics". J. Comput. Chem. 35 (17): 1317–1329. doi:10.1002/jcc.23613. PMID 24752427. S2CID 8788528.
- 1 2 G.B. Sushko; I.A. Solov'yov; A.V. Solov'yov (2016). "Molecular dynamics for irradiation driven chemistry: application to the FEBID process". Eur. Phys. J. D. 70 (10): 217. Bibcode:2016EPJD...70..217S. doi:10.1140/epjd/e2016-70283-5. S2CID 54844470.
- 1 2 I.A. Solov'yov; A.V. Korol; A.V. Solov'yov (2017). Multiscale Modeling of Complex Molecular Structure and Dynamics with MBN Explorer. Springer International Publishing. ISBN 978-3-319-56085-4.
- ↑ "DCD Trajectory I/O".
- ↑ "Protein Data Bank".
- ↑ G.B. Sushko; V.G. Bezchastnov; I.A. Solov'yov; A.V. Korol; W. Greiner; A.V. Solov'yov (2013). "Simulation of ultra-relativistic electrons and positrons channeling in crystals with MBN Explorer". J. Comput. Phys. 252: 404–418. arXiv: 1307.6771 . Bibcode:2013JCoPh.252..404S. doi:10.1016/j.jcp.2013.06.028. S2CID 2157486.
- ↑ "About MBN Studio". mbnresearch.com. Retrieved 31 August 2017.
- ↑ "FP7 ITN ARGENT Project".
- ↑ "PEARL Project".
- ↑ "COST Action Nano-IBCT".
- ↑ "VINAT Project".