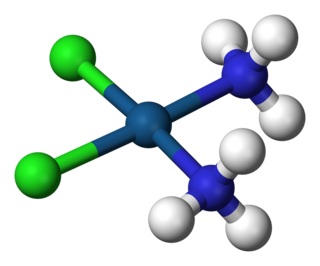
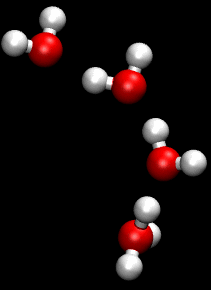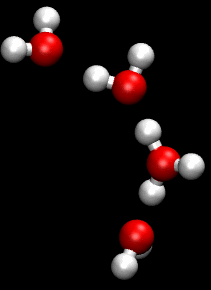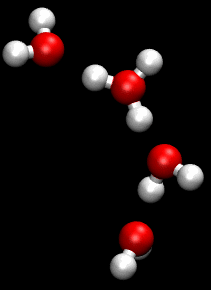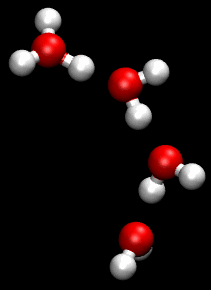
Properties and behavior of hydrated cations in aqueous solution
A metal ion in aqueous solution or aqua ion is a cation, dissolved in water, of chemical formula [M(H2O)n]z+. The solvation number, n, determined by a variety of experimental methods is 4 for Li+ and Be2+ and 6 for most elements in periods 3 and 4 of the periodic table. Lanthanide and actinide aqua ions have higher solvation numbers (often 8 to 9), with the highest known being 11 for Ac3+. The strength of the bonds between the metal ion and water molecules in the primary solvation shell increases with the electrical charge, z, on the metal ion and decreases as its ionic radius, r, increases. Aqua ions are subject to hydrolysis. The logarithm of the first hydrolysis constant is proportional to z2/r for most aqua ions.
The aqua ion is associated, through hydrogen bonding with other water molecules in a secondary solvation shell. Water molecules in the first hydration shell exchange with molecules in the second solvation shell and molecules in the bulk liquid. The residence time of a molecule in the first shell varies among the chemical elements from about 100 picoseconds to more than 200 years. Aqua ions are prominent in electrochemistry.
* No experimental information regarding aqua ion structures
Most chemical elements are metallic. Compounds of the metallic elements usually form simple aqua ions with the formula [M(H2O)n]z+ in low oxidation states. With the higher oxidation states the simple aqua ions dissociate losing hydrogen ions to yield complexes that contain both water molecules and hydroxide or oxide ions, such as the vanadium(IV) species [VO(H2O)5]2+. In the highest oxidation states only oxyanions, such as the permanganate(VII) ion, MnO− 4, are known. A few metallic elements that are commonly found only in high oxidation states, such as niobium and tantalum, are not known to form aqua cations; near the metal–nonmetal boundary, arsenic and tellurium are only known as hydrolysed species. Some elements, such as tin and antimony, are clearly metals, but form only covalent compounds in the highest oxidation states: their aqua cations are restricted to their lower oxidation states.[1]Germanium is a semiconductor rather than a metal, but appears to form an aqua cation; similarly, hydrogen forms an aqua cation like metals, despite being a gas. The transactinides have been greyed out due to a lack of experimental data. For some highly radioactive elements, experimental chemistry has been done, and aqua cations may have been formed, but no experimental information is available regarding the structure of those putative aqua ions.
Schematic representation of the aqua ion [Na(H2O)6] . The oxygen atoms are arranged at the vertices of a regular octahedron centered on the sodium ion.First and second solvation shells of an octahedral aqua ion. Up to 12 water molecules may be present in the second shell (only two are shown in this diagram) linked by hydrogen bonds to the molecules in the first shell.
In aqueous solution the water molecules directly attached to the metal ion are said to belong to the first coordination sphere, also known as the first, or primary, solvation shell. The bond between a water molecule and the metal ion is a dative covalent bond, with the oxygen atom donating both electrons to the bond. Each coordinated water molecule may be attached by hydrogen bonds to other water molecules. The latter are said to reside in the second coordination sphere. The second coordination sphere is not a well defined entity for ions with charge 1 or 2. In dilute solutions it merges into the water structure in which there is an irregular network of hydrogen bonds between water molecules.[2] With tripositive ions the high charge on the cation polarizes the water molecules in the first solvation shell to such an extent that they form strong enough hydrogen bonds with molecules in the second shell to form a more stable entity.[3]
The strength of the metal-oxygen bond can be estimated in various ways. The hydration enthalpy, though based indirectly on experimental measurements, is the most reliable measure. The scale of values is based on an arbitrarily chosen zero, but this does not affect differences between the values for two metals. Other measures include the M–O vibration frequency and the M–O bond length. The strength of the M-O bond tends to increase with the charge and decrease as the size of the metal ion increases. In fact there is a very good linear correlation between hydration enthalpy and the ratio of charge squared to ionic radius, z2/r.[4] For ions in solution Shannon's "effective ionic radius" is the measure most often used.[5]
Water molecules in the first and second solvation shells can exchange places. The rate of exchange varies enormously, depending on the metal and its oxidation state. Metal aqua ions are always accompanied in solution by solvated anions, but much less is known about anion solvation than about cation solvation.[6]
Many other aqua ions are present in seawater in concentrations ranging from ppm to ppt.[9] The concentrations of sodium, potassium, magnesium and calcium in blood are similar to those of seawater. Blood also has lower concentrations of essential elements such as iron and zinc. Sports drink is designed to be isotonic and also contains the minerals which are lost in perspiration.
Magnesium and calcium ions are common constituents of domestic water and are responsible for permanent and temporary hardness, respectively. They are often found in mineral water.
Experimental methods
Information obtained on the nature of ions in solution varies with the nature of the experimental method used. Some methods reveal properties of the cation directly, others reveal properties that depend on both cation and anion. Some methods supply information of a static nature, a kind of snapshot of average properties, others give information about the dynamics of the solution.
Nuclear magnetic resonance (NMR)
Ions for which the water-exchange rate is slow on the NMR time-scale give separate peaks for molecules in the first solvation shell and for other water molecules. The solvation number is obtained as a ratio of peak areas. Here it refers to the number of water molecules in the first solvation shell. Molecules in the second solvation shell exchange rapidly with solvent molecules, giving rise to a small change in the chemical shift value of un-coordinated water molecules from that of water itself. The main disadvantage of this method is that it requires fairly concentrated solutions, with the associated risk of ion-pair formation with the anion.
A solution containing an aqua ion does not have the long-range order that would be present in a crystal containing the same ion, but there is short-range order. X-ray diffraction on solutions yields a radial distribution function from which the coordination number of the metal ion and metal-oxygen distance may be derived. With aqua ions of high charge some information is obtained about the second solvation shell.[11][12]
This technique requires the use of relatively concentrated solutions. X-rays are scattered by electrons, so scattering power increases with atomic number. This makes hydrogen atoms all but invisible to X-ray scattering.
Large angle X-ray scattering has been used to characterize the second solvation shell with trivalent ions such as Cr3+ and Rh3+. The second hydration shell of Cr3+ was found to have 13±1 molecules at an average distance of 402±20pm. This implies that every molecule in the first hydration shell is hydrogen bonded to two molecules in the second shell.[13]
Neutron diffraction
Diffraction by neutrons also give a radial distribution function. In contrast to X-ray diffraction, neutrons are scattered by nuclei and there is no relationship with atomic number.[14] Indeed, use can be made of the fact that different isotopes of the same element can have widely different scattering powers. In a classic experiment, measurements were made on four nickel chloride solutions using the combinations of 58Ni, 60Ni, 35Cl and 37Cl isotopes to yield a very detailed picture of cation and anion solvation.[15] Data for a number of metal salts show some dependence on the salt concentration.
Cation hydration in solution, determined by neutron diffraction[16]
Salt
LiCl
CaCl2
NiCl2
Molality of salt
27.77
9.95
3.57
4.49
2.80
1.0
3.05
0.85
0.46
0.086
Cation hydration number†
2.3 (2)
3.0 (5)
5.5 (3)
6.4 (3)
7.2 (2)
10.0 (6)
5.8 (2)
6.6 (5)
6.8 (8)
6.8 (8)
θ /deg‡
75 (5)
52 (5)
40 (5)
34 (9)
34 (9)
38 (9)
42 (8)
27 (10)
17 (10)
0 (20)
(continued)
Salt
Ni(ClO4)2
Cu(ClO4)2
Fe(NO3)3
NdCl3
DyCl3
Molality of salt
3.80
2.00
2.0
2.85
2.38
Cation hydration number†
5.8 (2)
4.9 (3)
5.0 (2)
8.5 (2)
7.4 (5)
θ /deg‡
42 (8)
38 (6)
22 (4)
24 (4)
17 (3)
†Figures in brackets are standard deviations on the last significant figure of the value.‡ angle between a M-OH2 bond and the plane of the water molecule.
Most of these data refer to concentrated solutions in which there are very few water molecules that are not in the primary hydration spheres of the cation or anion, which may account for some of the variation of solvation number with concentration even if there is no contact ion pairing. The angle θ gives the angle of tilt of the water molecules relative to a plane in the aqua ion. This angle is affected by the hydrogen bonds formed between water molecules in the primary and secondary solvation shells.
The measured solvation number is a time-averaged value for the solution as a whole. When a measured primary solvation number is fractional there are two or more species with integral solvation numbers present in equilibrium with each other. This also applies to solvation numbers that are integral numbers, within experimental error. For example, the solvation number of 5.5 for a lithium chloride solution could be interpreted as being due to presence of two different aqua ions with equal concentrations.
[Li(H2O)6]+⇌ [Li(H2O)5]+ + H2O
Another possibility is that there is interaction between a solvated cation and an anion, forming an ion pair. This is particularly relevant when measurements are made on concentrated salt solutions. For example, a solvation number of 3 for a lithium chloride solution could be interpreted as being due to the equilibrium
[Li(H2O)4]+ + Cl−⇌ [Li(H2O)3Cl] + H2O
lying wholly in favour of the ion pair.
Vibrational spectra
Infrared spectra and Raman spectra can be used to measure the M-O stretching frequency in metal aqua ions. Raman spectroscopy is particularly useful because the Raman spectrum of water is weak whereas the infrared spectrum of water is intense. Interpretation of the vibration frequencies is somewhat complicated by the presence, in octahedral and tetrahedral ions, of two vibrations, a symmetric one measured in the Raman spectrum and an anti-symmetric one, measured in the infrared spectrum.
Symmetric M-O stretching vibrations of some aqua ions in solution [17][18]
Although the relationship between vibration frequency and force constant is not simple, the general conclusion that can be taken from these data is that the strength of the M-O bond increases with increasing ionic charge and decreasing ionic size. The M-O stretching frequency of an aqua ion in solution may be compared with its counterpart in a crystal of known structure. If the frequencies are very similar it can be concluded that the coordination number of the metal ion is the same in solution as it is in a compound in the solid state.
Dynamic methods
Data such as conductivity, electrical mobility and diffusion relate to the movement of ions through a solution. When an ion moves through a solution it tends to take both first and second solvation shells with it. Hence solvation numbers measured from dynamic properties tend to be much higher that those obtained from static properties.
Hydrogen is not a metal, but like them it tends to lose its valence electron in chemical reactions, forming a cation H+. In aqueous solution, this immediately attaches itself to a water molecule,[20] forming a species generally symbolised as H3O+ (sometimes loosely written H+). Such hydration forms cations that can in essence be considered as [H(OH2)n]+.[21]
The solvation of H+ in water is not fully characterised and many different structures have been suggested. Two well-known structures are the Zundel cation and the Eigen cation. The Eigen solvation structure has the hydronium ion at the center of an H9O+4 complex in which the hydronium is strongly hydrogen-bonded to three neighbouring water molecules. In the Zundel H5O+2 complex the proton is shared equally by two water molecules in a symmetric hydrogen bond.[22][23][24][25][26]
Alkali metals
The hydrated lithium cation in water is probably tetrahedral and four-coordinated.[27] There are most probably six water molecules in the primary solvation sphere of the octahedral sodium ion.[27][28]Potassium is seven-coordinate, and rubidium and caesium are probably eight-coordinate square antiprismatic.[27] No data is available for francium.
Alkaline earth metals
Group 2 cations in aqueous solution
[Be(H2O)4]2+
[Mg(H2O)6]2+
Ca2+(aq)
Sr2+(aq)
Ba2+(aq)
M-O distance (pm)
167
209
242§
263§
281§
solvation (kJ mol−1)
2494
1921
1577
1443
1305
§ Values extrapolated from data for solid-state crystal structures
The beryllium cation [Be(H2O)4]2+ has a very well-defined primary solvation shell with a tetrahedral BeO4 core.[29] For magnesium, [Mg(H2O)6]2+ is also a well-characterized species, with an octahedral MgO6 core.[29] The situation for calcium is more complicated. Neutron diffraction data gave a solvation number for calcium chloride, CaCl2, which is strongly dependent on concentration: 10.0±0.6 at 1mol·dm−3, decreasing to 6.4±0.3 at 2.8mol·dm−3. The enthalpy of solvation decreases with increasing ionic radius. Various solid hydrates are known with 8-coordination in square antiprism and dodecahedral geometry.[30] In water, calcium and strontium are most probably eight-coordinate square antiprismatic (although seven-coordination for calcium cannot presently be excluded). Barium is not as well-studied: it seems to have a coordination number of either eight or nine. Theoretical simulation of radium suggests that its aqua cation is ten-coordinate.[27]
Group 3 metals, lanthanides and actinides
face-capped trigonal prism structure
Scandium(III) and yttrium(III) are both eight-coordinate, but have different structures: scandium has an unusual dicapped triangular prismatic structure (with one cap location empty), while yttrium is square antiprismatic. Lutetium(III) is tricapped triangular prismatic, but has a significant water deficit: one of the capping water molecules is significantly closer to the lutetium than the remaining ones and the average coordination number is only 8.2 rather than 9. Based on its ionic radius, lawrencium(III) is probably nine-coordinate tricapped triangular prismatic with no water deficit.[27]
The trivalent lanthanide ions decrease steadily in size from lanthanum to lutetium, an effect known as the lanthanide contraction.[31] From lanthanum to dysprosium, the coordination number is maintained at 9 with a tricapped trigonal prismatic structure, although starting from samarium the capping water molecules are no longer equally strongly bounded. A water deficit then appears for holmium through lutetium with the average coordination number dropping to 8.2 at lutetium(III). The configuration is maintained despite the small size of the cations and the water deficit, probably due to strong hydrogen bonding.[32]Europium(II) is seven-coordinate, and cerium(IV) is hydrolysed to the oxygen-bridged dimer [(H2O)7Ce–O–Ce(OH2)7]6+.[27]
Actinium(III) is eleven-coordinate in aqueous solution. Thorium(IV) is nine-coordinate tricapped trigonal prismatic, and it is assumed that the same is true for the other actinide(IV) cations in aqueous solutions (as that is also their solid-state configuration). Studies on coordination number and/or structure for actinides(III) to date stretch only to californium.[32][33] However, since lawrencium(III) has a similar ionic radius to dysprosium(III), it is likely that uranium(III) through lawrencium(III) are all nine-coordinate tricapped triangular prismatic with the capping positions fully occupied.[32][27] No data is available for fermium(II), mendelevium(II), or nobelium(II).
Jahn-Teller distorted octahedral structure of [Cu(H2O)6] found in the solid state and possibly also in solutionProposed square pyramidal structure of [Cu(H2O)5] in aqueous solution
The ions of these metals in the +2 and +3 oxidation states have a solvation number of 6. All have a regular octahedral structure except the aqua ions of chromium(II) and copper(II) which are subject to Jahn-Teller distortion. In the copper case the two axial Cu−O distances are 238 pm, whereas the four equatorial Cu−O distances are 195 pm in the solid state.[35] However, it is unclear whether Cu2+ has a solvation number of 5 or 6 in aqueous solution, with conflicting experimental reports.[32] The structure of cobalt(III) in aqueous solution has not been determined.[27] Copper(I) is estimated to be four-coordinate tetrahedral.[27]
A solvation number of 6 with an octahedral structure is well established for zinc(II) and cadmium(II) in dilute solutions. In concentrated solutions the Zn2+ ion may adopt a 4-coordinate, tetrahedral, structure, but the evidence is not conclusive because of the possibility of ion pairing and/or hydrolysis.[36] The solvation number of mercury(II) is most likely to be 6.[37]Zinc(II) is six-coordinate octahedral, but cadmium(II) may be in equilibrium between six- and seven-coordination. Mercury(II) is a pseudo-Jahn-Teller-distorted octahedron.[27] The bis aqua structure of the mercury(I) ion, [(H2O)-Hg-Hg-(OH2)]+, found in solid compounds,[38] is not the same as that found in solution which involves three water molecules coordinated to each mercury completing a distorted tetrahedral arrangement.[27] Another aqua species in which there is a metal-metal bond is the molybdenum(II) species formulated as [(H2O)4Mo≣Mo(H2O)4]4+.[39] Each molybdenum is surrounded by four water molecules in a square-planar arrangement, in a structure similar to that of the known structure of the chloro complex [Mo2Cl8]4−.[40]
There are a few divalent and trivalent aqua ions of transition metals in the second and third transition series: ruthenium(II) and (III), rhodium(III), and iridium(III), all octahedral. (Ruthenium and iridium structures have only been examined in the solid state, but it is assumed that they are the same in aqueous solution.)[27]Molybdenum(III) is questionable (and may be strongly hydrolyzed in aqueous solution), and molybdenum(II) dimerises with each molybdenum binding four water molecules.[27][32]Palladium(II) and platinum(II) aqua ions were originally thought to be square planar, but are actually strongly tetragonally elongated square-pyramidal or octahedral with the extra one or two water molecules extremely loosely bound.[27] The structure of silver(I) is disputed: it may be two-coordinate, or it may be four-coordinate with two extra very loosely bound water molecules.[27]Gold(III) is four-coordinate square planar in the solid state, and it is assumed to have the same structure in aqueous solution.[27] Distortion occurs for low-coordinate metals with strong covalent tendencies due to the second-order Jahn-Teller effect. With oxidation state 4, however, the only unhydrolyzed species are the square antiprismatic zirconium(IV), [Zr(H2O)8]4+, and hafnium(IV), [Hf(H2O)8]4+, and even they are extremely prone to hydrolysis.[32] Such a zirconium cation is only formed in dilute solutions of ZrIV in strong acid, and in practice the cationic species encountered of zirconium and hafnium are polynuclear.[41]
Group 13-18 elements
Boron is not a metal, and boron(III) is too acidic for an aqua ion to exist: deprotonation proceeds as far as boric acid, borates, and hydroxyborates.[42] The aluminium(III) aqua ion, [Al(H2O)6]3+ is very well characterized in solution and the solid state. The AlO6 core has octahedral symmetry, point group Oh. The aqua ions of gallium(III), indium(III) and thallium(III) are also six-coordinate octahedral.[27] The coordination geometry of thallium(I) is not experimentally known, but it is likely to be hemidirected with a large gap in the coordination sphere.[27]
Silicon is likewise not a metal, and silicon(IV) is a strong enough acid to deprotonate bound OH−. Thus various forms of hydrated silica (silicic acid) form.[43] There is some evidence that germanium(II) aqua ions can form in perchloric acid media.[44] Quantum mechanical calculations suggests that the germanium(II) aqua ion shows extreme distortion of the first coordination sphere due to the high charge density and the stereochemically active lone pairs. The first shell is calculated to usually have a solvation number of 6, but numbers 4–7 are also possible and the shell splits into two with differing distances from the central Ge2+.[45] However, germanium(II) is readily oxidised to germanium(IV),[46] for which only hydrolyzed species are expected.[47] The important germanium(IV) species are anionic oxo-hydroxo mixed species, thus displaying intermediate behaviour between silicon and tin: the major species appear to be [GeO(OH)3]− and the octameric [Ge8O16(OH)3]3−, with [GeO2(OH)2]2− occurring in smaller quantities.[43]Tin(II) is 3-coordinate hemidirected[48][49] with a very large gap in the coordination sphere of tin(II).[27] The hydration number of lead(II) is not well-established and could be anywhere from five to seven.[27] In practice these cations tend to be polynuclear.[46] For tin(IV) and lead(IV) there are only hydrolyzed species.[47]
Arsenic(III) is calculated to form hydrolyzed species only.[47] The stable cationic arsenic(III) species in water is calculated to be [As(OH)2]+,[50] though hydrolysis usually proceeds further to neutral and anionic species.[51]Antimony(III) aqua ions may exist in dilute solutions of antimony(III) in concentrated acids.[51][52] Quantum mechanical calculations reveal a solvation number of 8, with the first coordination sphere splitting into two hydration hemispheres with 4 water molecules each.[53]Bismuth(III) is eight-coordinate square antiprismatic in aqueous solution, though in the solid state it is nine-coordinate tricapped triangular prismatic.[27] Although the structures for thallium(I), germanium(II), tin(II), lead(II), and antimony(III) are affected by the lone pairs, this is not so for bismuth(III).[32]
Selenium(IV) is mostly present as selenous acid (H2SeO3) below pH 2; at higher pH this deprotonates to HSeO3− and then SeO32−.[54] Cationic tellurium(IV) appears to be [Te(OH)3]+; it predominates in dilute solutions below pH 2. Above pH 4, the dominating species becomes TeO(OH)3−, and above pH 8 it becomes TeO2(OH)22−.[55]Polonium(IV) should be similar to tellurium(IV), though a little weaker, in its tendency towards hydrolysis.[56] The structure of polonium(II) does not appear to have been studied.
The halogens, being strongly nonmetallic, prefer to form anions rather than cations in aqueous solution.[57] Anion solvation is complicated because the water molecules point the other way: cations bind to the oxygen atom of water, with the hydrogens facing away, while anions prefer to bond asymmetrically to only one of the hydrogen atoms in a nearby water molecule. This results in significant water–water hydrogen bonding and network formation already within the first hydration shell, to an extent that does not occur for cation solvation. Such interactions are larger for the heavier and larger halides; the hydrogen bonding decreases in strength as one proceeds from iodide to fluoride, because of increasing negative charge on the water molecules, the increasing inductive effect stemming from the higher electric fields, and increasing geometrical strain for the hydrogen bonding.[58] The rare and extremely radioactive astatine seems to be more metallic: a cationic astatine(I) species is inferred from trace-scale experiments in acidic solutions, and sometimes symbolised At+, but its structure has not been determined.[59]
The noble gases do not react with water, but their solubility in water increases when going down the group. Argon atoms in water appear to have a first hydration shell composed of 16±2 water molecules at a distance of 280–540pm, and a weaker second hydration shell is found out to 800pm. Similar hydration spheres have been found for krypton and xenon atoms in water.[60]
Oxo-aqua-cations
Some elements in oxidation states higher than 3 form stable, aquated, oxo ions. Well known examples are the vanadyl(IV) and uranyl(VI) ions. They can be viewed as particularly stable hydrolysis products in a hypothetical reaction such as
[V(H2O)6]4+ → [VO(H2O)5]2+ + 2H+
The vanadium has a distorted octahedral environment (point group C4v) of one oxide ion and 5 water molecules.[61] Titanyl, TiO2+, has a similar structure.[32] Vanadium(V) is believed to exist as the dioxo-ion [VO2(H2O)4]+ at pH less than 2, but the evidence for this ion depends on the formation of complexes, such as oxalate complexes which have been shown to have the VO+ 2 unit, with cis-VO bonds, in the solid state.[62] The chromium(IV) ion [CrO(H2O)5]2+, similar to the vanadium ion has been proposed on the basis of indirect evidence.[63]
The uranyl ion, UO2+ 2, has a trans structure. The aqua ion UO2+ 2(aq) has five water molecules in the plane perpendicular to the O-U-O axis in a pentagonal bipyramid structure, point group D5h. Neptunyl and plutonyl have the same structure. Nothing is known of actinide(V) structures.[27]
Thermodynamics
The main goal of thermodynamics in this context is to derive estimates of single-ion thermodynamic quantities such as hydration enthalpy and hydration entropy. These quantities relate to the reaction
Mz+ (gas) + solvent → Mz+ (in solution)
The enthalpy for this reaction is not directly measurable, because all measurements use salt solutions that contain both cation and anion. Most experimental measurements relate to the heat evolved when a salt dissolves in water, which gives the sum of cation and anion solvation enthalpies. Then, by considering the data for different anions with the same cation and different cations with the same anion, single ion values relative to an arbitrary zero, are derived.
Minus hydration enthalpy for (octahedral) divalent transition metal M ionsHydration enthalpies of trivalent lanthanide Ln ions
Single ion standard hydration enthalpy /kJ mol−1[64]
Li+ -514.6
Be2+ -2487.0
Na+ -404.6
Mg2+ -1922.1
Al3+ -4659.7
K+ -320.9
Ca2+ -1592.4
Sc3+ -3960.2
...
Ga3+ -4684.8
Rb+ -296.2
Sr2+ -1444.7
Y3+ -3620.0
...
In3+ -4108.7
Sn2+ -1554.4
Cs+ -263.2
Ba2+ -1303.7
La3+ -3282.8
...
Tl3+ -4184.0
Pb2+ -1479.9
Other values include Zn2+ -2044.3, Cd2+ -1805.8 and Ag+ -475.3 kJ mol−1.
There is an excellent linear correlation between hydration enthalpy and the ratio of charge squared, z2, to M-O distance, reff.[65]
Values for transition metals are affected by crystal field stabilization. The general trend is shown by the magenta line which passes through Ca2+, Mn2+ and Zn2+, for which there is no stabilization in an octahedral crystal field. Hydration energy increases as size decreases. Crystal field splitting confers extra stability on the aqua ion. The maximum crystal field stabilization energy occurs at Ni2+. The agreement of the hydration enthalpies with predictions provided one basis for the general acceptance of crystal field theory.[66]
The hydration enthalpies of the trivalent lanthanide ions show an increasingly negative values at atomic number increases, in line with the decrease in ionic radius known as the lanthanide contraction.
Single ion hydration entropy can be derived. Values are shown in the following table. The more negative the value, the more there is ordering in forming the aqua ion. It is notable that the heavy alkali metals have rather small entropy values which suggests that both the first and second solvation shells are somewhat indistinct.
Single ion standard hydration entropy at 25°C /J deg−1 mol−1[67]
There are two ways of looking at an equilibrium involving hydrolysis of an aqua ion. Considering the dissociation equilibrium
[M(H2O)n]z+ - H+⇌ [M(H2O)n-1(OH)](z-1)+
the activity of the hydrolysis product, omitting the water molecules, is given by
The alternative is to write the equilibrium as a complexation or substitution reaction
[M(H2O)n]z+ +OH−⇌:[M(H2O)n-1(OH)](z-1)+ + H2O
In which case
The concentration of hydrogen and hydroxide ions are related by the self-ionization of water, Kw = {H+} {OH−} so the two equilibrium constants are related as
In practice the first definition is more useful because equilibrium constants are determined from measurements of hydrogen ion concentrations. In general,
charges are omitted for the sake of generality and activities have been replaced by concentrations. are cumulative hydrolysis constants.
Modeling the hydrolysis reactions that occur in solution is usually based on the determination of equilibrium constants from potentiometric (pH) titration data. The process is far from straightforward for a variety of reasons.[68] Sometimes the species in solution can be precipitated as salts and their structure confirmed by X-ray crystallography. In other cases, precipitated salts bear no relation to what is postulated to be in solution, because a particular crystalline substances may have both low solubility and very low concentration in the solutions.
First hydrolysis constant
The logarithm of hydrolysis constant, K1,-1, for the removal of one proton from an aqua ion
[M(H2O)n]z+ - H+⇌ [M(H2O)n-1(OH)](z-1)+
[ [M(OH)]{(z-1)+ ] = K1,-1 [Mz+] [H+] −1
shows a linear relationship with the ratio of charge to M-O distance, z/d. Ions fall into four groups. The slope of the straight line is the same for all groups, but the intercept, A, is different.[69]
The cations most resistant to hydrolysis for their size and charge are hard pre-transition metal ions or lanthanide ions. The slightly less resistant group includes the transition metal ions. The third group contains mostly soft ions ion of post-transition metals. The ions which show the strongest tendency to hydrolyze for their charge and size are Pd2+, Sn2+ and Hg2+.[69] This is because of the low coordination numbers of ions in this part of the periodic table (also including Ag+ and Au+), so that fewer water molecules are present around the cation and they experience more electrostatic force than normal. A similar situation affects Be2+, the smallest aqua cation, which is also more acidic than would normally be expected.[70]
The standard enthalpy change for the first hydrolysis step is generally not very different from that of the dissociation of pure water. Consequently, the standard enthalpy change for the substitution reaction
[M(H2O)n]z+ +OH−⇌:[M(H2O)n-1(OH)](z-1)+ + H2O
is close to zero. This is typical of reactions between a hard cation and a hard anion, such as the hydroxide ion.[71] It means that the standard entropy charge is the major contributor to the standard free energy change and hence the equilibrium constant.
The change in ionic charge is responsible for the effect as the aqua ion has a greater ordering effect on the solution than the less highly charged hydroxo complex.
Multiple hydrolysis reactions
Beryllium hydrolysis as a function of pH. Water molecules attached to Be are omittedtrimeric hydrolysis product of berylliumThe molybdenum(IV) oxo-aqua ion [Mo3O4(H2O)9]
The hydrolysis of beryllium shows many of the characteristics typical of multiple hydrolysis reactions. The concentrations of various species, including polynuclear species with bridging hydroxide ions, change as a function of pH up to the precipitation of an insoluble hydroxide. Beryllium hydrolysis is unusual in that the concentration of [Be(H2O)3(OH)]+ is too low to be measured. Instead a trimer ([Be3(H2O)6(OH3))3+ is formed, whose structure has been confirmed in solid salts. The formation of polynuclear species is driven by the reduction in charge density within the molecule as a whole. The local environment of the beryllium ions approximates to [Be(H2O)2(OH)2]+. The reduction in effective charge releases free energy in the form of a decrease of the entropy of ordering at the charge centers.[72]
six-membered ring with alternate Mn+ and OH− groups
M 3(OH)(3z-4)+ 4
Sn2+, Pb2+ Al3+, Cr3+, Fe3+, In3+
Cube with alternate vertices of Mn+ and OH− groups, one vertex missing
M 4(OH)4+ 4
Mg2+, Co2+, Ni2+, Cd2+, Pb2+
Cube with alternate vertices of Mn+ and OH− groups
M 4(OH)8+ 8
Zr4+, Th4+
Square of Mn+ ions with double hydroxide bridges on each side of the square
The hydrolysis product of aluminium formulated as [Al13O4(OH)24(H2O)12]7+ is very well characterized and may be present in nature in water at pH ca. 5.4.[74]
The overall reaction for the loss of two protons from an aqua ion can be written as
[M(H2O)n]z+ - 2 H+⇌ [M(H2O)n-2(OH)2](z-2)+
However, the equilibrium constant for the loss of two protons applies equally well to the equilibrium
[M(H2O)n]z+ - 2 H+⇌ [MO(H2O)n-2](z-2)+ + H2O
because the concentration of water is assumed to be constant. This applies in general: any equilibrium constant is equally valid for a product with an oxide ion as for the product with two hydroxyl ions. The two possibilities can only be distinguished by determining the structure of a salt in the solid state. Oxo bridges tend to occur when the metal oxidation state is high.[75] An example is provided by the molybdenum(IV) complex [Mo3O4(H2O)9]4+ in which there is a triangle of molybdenum atoms joined by σ- bonds with an oxide bridge on each edge of the triangle and a fourth oxide which bridges to all three Mo atoms.[76]
There are very few oxo-aqua ions of metals in the oxidation state +5 or higher. Rather, the species found in aqueous solution are monomeric and polymeric oxyanions. Oxyanions can be viewed as the end products of hydrolysis, in which there are no water molecules attached to the metal, only oxide ions.
Exchange kinetics
A water molecule in the first solvation shell of an aqua ion may exchange places with a water molecule in the bulk solvent. It is usually assumed that the rate-determining step is a dissociation reaction.
[M(H2O)n]z+ → [M(H2O)n-1]z+* + H2O
The * symbol signifies that this is the transition state in a chemical reaction. The rate of this reaction is proportional to the concentration of the aqua ion, [A].
.
The proportionality constant, k, is called a first-order rate constant at temperature T. The unit of the reaction rate for water exchange is usually taken as mol dm−3s−1.
The half-life for this reaction is equal to loge2 / k. This quantity with the dimension of time is useful because it is independent of concentration. The quantity 1/k, also with dimension of time, equal to the half life divided by 0.6932, is known as the residence time or time constant.[77]
The residence time for water exchange varies from about 10−10s for Cs+ to about 10+10 s (more than 200 y) for Ir3+. It depends on factors such as the size and charge on the ion and, in the case of transition metal ions, crystal field effects. Very fast and very slow reactions are difficult to study. The most information on the kinetics a water exchange comes from systems with a residence time between about 1 μs and 1 s. The enthalpy and entropy of activation, ΔH‡ and ΔS‡ can be obtained by observing the variation of rate constant with temperature.
Kinetic parameters (at 25°C) for water exchange: divalent ions, M2+ (aq) [78]
Be
Mg
V
Cr
Mn
Fe
Co
Ni
Cu
Zn
UO2
Residence time (μs)
0.001
2
0.00013
0.0032
0.0316
0.32
0.79
40
0.0005
0.032
1.3
ΔH‡ (kJ mol−1)
43
69
13
34
32
33
43
23
ΔS‡ (J deg−1mol−1)
8
21
-13
12
-13
-17
-22
25
Note the general increase in the residence time from vanadium to nickel, which mirrors the decrease in ion size with increasing atomic number, which is a general trend in the periodic table, though given a specific name only in the case of the lanthanide contraction. The effects of crystal field stabilization energy are superimposed on the periodic trend.
Kinetic parameters (at 25°C) for water exchange - trivalent ions, M3+ (aq)[78]
Al
Ti
Cr
Fe
Ga
Rh
In
La
residence time (μs)
6.3×106
16
2.0×1012
316
501
3.2×1013
50
0.050
ΔH‡ (kJ mol−1)
11
26
109
37
26
134
17
ΔS‡ (J deg−1mol−1)
117
-63
0
-54
-92
59
Solvent exchange is generally slower for trivalent than for divalent ions, as the higher electrical charge on the cation makes for stronger M-OH2 bonds and, in consequence, higher activation energy for the dissociative reaction step, [M(H2O)n]3+ → [M(H2O)n-1]3+ + H2O. The values in the table show that this is due to both activation enthalpy and entropy factors.[79]
The ion [Al(H2O)6]3+ is relatively inert to substitution reactions because its electrons are effectively in a closed shell electronic configuration, [Ne]3s23p6, making dissociation an energy-expensive reaction. Cr3+, which has an octahedral structure and a d3 electronic configuration is also relatively inert, as are Rh3+ and Ir3+ which have a low-spin d6 configuration.
Metal aqua ions are often involved in the formation of complexes. The reaction may be written as
pMx+(aq) + qLy− → [MpLq](px-qy)+
In reality this is a substitution reaction in which one or more water molecules from the first hydration shell of the metal ion are replaced by ligands, L. The complex is described as an inner-sphere complex. A complex such as [ML](p-q)+ may be described as a contact ion pair.
When the water molecule(s) of the second hydration shell are replaced by ligands, the complex is said to be an outer-sphere complex, or solvent-shared ion pair. The formation of solvent-shared or contact ion pairs is particularly relevant to the determination of solvation numbers of aqua ions by methods that require the use of concentrated solutions of salts, as ion pairing is concentration-dependent. Consider, for example, the formation of the complex [MgCl]+ in solutions of MgCl2. The formation constant K of the complex is about 1 but varies with ionic strength.[80] The concentration of the rather weak complex increases from about 0.1% for a 10mM solution to about 70% for a 1M solution (1M = 1mol dm−3).
Electrochemistry
The standard electrode potential for the half-cell equilibrium Mz+ + z e−⇌ M(s) has been measured for all metals except for the heaviest trans-uranium elements.
As the standard electrode potential is more negative the aqua ion is more difficult to reduce. For example, comparing the potentials for zinc (-0.75 V) with those of iron (Fe(II) -0.47 V, Fe(III) -0.06 V) it is seen that iron ions are more easily reduced than zinc ions. This is the basis for using zinc to provide anodic protection for large structures made of iron or to protect small structures by galvanization.
Related Research Articles
A coordination complex is a chemical compound consisting of a central atom or ion, which is usually metallic and is called the coordination centre, and a surrounding array of bound molecules or ions, that are in turn known as ligands or complexing agents. Many metal-containing compounds, especially those that include transition metals, are coordination complexes.
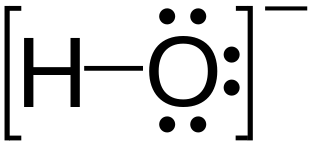
Hydroxide is a diatomic anion with chemical formula OH−. It consists of an oxygen and hydrogen atom held together by a single covalent bond, and carries a negative electric charge. It is an important but usually minor constituent of water. It functions as a base, a ligand, a nucleophile, and a catalyst. The hydroxide ion forms salts, some of which dissociate in aqueous solution, liberating solvated hydroxide ions. Sodium hydroxide is a multi-million-ton per annum commodity chemical. The corresponding electrically neutral compound HO• is the hydroxyl radical. The corresponding covalently bound group –OH of atoms is the hydroxy group. Both the hydroxide ion and hydroxy group are nucleophiles and can act as catalysts in organic chemistry.
Hydrolysis is any chemical reaction in which a molecule of water breaks one or more chemical bonds. The term is used broadly for substitution, elimination, and solvation reactions in which water is the nucleophile.
Solvation describes the interaction of a solvent with dissolved molecules. Both ionized and uncharged molecules interact strongly with a solvent, and the strength and nature of this interaction influence many properties of the solute, including solubility, reactivity, and color, as well as influencing the properties of the solvent such as its viscosity and density. If the attractive forces between the solvent and solute particles are greater than the attractive forces holding the solute particles together, the solvent particles pull the solute particles apart and surround them. The surrounded solute particles then move away from the solid solute and out into the solution. Ions are surrounded by a concentric shell of solvent. Solvation is the process of reorganizing solvent and solute molecules into solvation complexes and involves bond formation, hydrogen bonding, and van der Waals forces. Solvation of a solute by water is called hydration.
In chemistry, hydronium (hydroxonium in traditional British English) is the common name for the cation [H3O]+, also written as H3O+, the type of oxonium ion produced by protonation of water. It is often viewed as the positive ion present when an Arrhenius acid is dissolved in water, as Arrhenius acid molecules in solution give up a proton (a positive hydrogen ion, H+) to the surrounding water molecules (H2O). In fact, acids must be surrounded by more than a single water molecule in order to ionize, yielding aqueous H+ and conjugate base. Three main structures for the aqueous proton have garnered experimental support: the Eigen cation, which is a tetrahydrate, H3O+(H2O)3, the Zundel cation, which is a symmetric dihydrate, H+(H2O)2, and the Stoyanov cation, an expanded Zundel cation, which is a hexahydrate: H+(H2O)2(H2O)4. Spectroscopic evidence from well-defined IR spectra overwhelmingly supports the Stoyanov cation as the predominant form. For this reason, it has been suggested that wherever possible, the symbol H+(aq) should be used instead of the hydronium ion.
An aqueous solution is a solution in which the solvent is water. It is mostly shown in chemical equations by appending (aq) to the relevant chemical formula. For example, a solution of table salt, also known as sodium chloride (NaCl), in water would be represented as Na+(aq) + Cl−(aq). The word aqueous means pertaining to, related to, similar to, or dissolved in, water. As water is an excellent solvent and is also naturally abundant, it is a ubiquitous solvent in chemistry. Since water is frequently used as the solvent in experiments, the word solution refers to an aqueous solution, unless the solvent is specified.
Zinc chloride is the name of inorganic chemical compounds with the formula ZnCl2·nH2O, with n ranging from 0 to 4.5, forming hydrates. Zinc chloride, anhydrous and its hydrates are colorless or white crystalline solids, and are highly soluble in water. Five hydrates of zinc chloride are known, as well as four forms of anhydrous zinc chloride. This salt is hygroscopic and even deliquescent. Zinc chloride finds wide application in textile processing, metallurgical fluxes, and chemical synthesis. No mineral with this chemical composition is known aside from the very rare mineral simonkolleite, Zn5(OH)8Cl2·H2O.
In chemistry, water(s) of crystallization or water(s) of hydration are water molecules that are present inside crystals. Water is often incorporated in the formation of crystals from aqueous solutions. In some contexts, water of crystallization is the total mass of water in a substance at a given temperature and is mostly present in a definite (stoichiometric) ratio. Classically, "water of crystallization" refers to water that is found in the crystalline framework of a metal complex or a salt, which is not directly bonded to the metal cation.
Aluminium chloride, also known as aluminium trichloride, is an inorganic compound with the formula AlCl3. It forms a hexahydrate with the formula [Al(H2O)6]Cl3, containing six water molecules of hydration. Both the anhydrous form and the hexahydrate are colourless crystals, but samples are often contaminated with iron(III) chloride, giving them a yellow colour.
The Grotthuss mechanism is a model for the process by which an 'excess' proton or proton defect diffuses through the hydrogen bond network of water molecules or other hydrogen-bonded liquids through the formation and concomitant cleavage of covalent bonds involving neighboring molecules.
Iron(III) nitrate, or ferric nitrate, is the name used for a series of inorganic compounds with the formula Fe(NO3)3.(H2O)n. Most common is the nonahydrate Fe(NO3)3.(H2O)9. The hydrates are all pale colored, water-soluble paramagnetic salts.
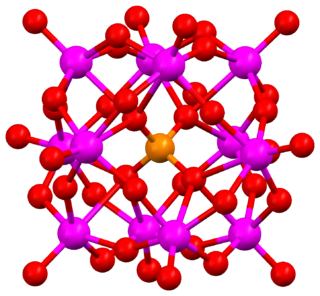
The Keggin structure is the best known structural form for heteropoly acids. It is the structural form of α-Keggin anions, which have a general formula of [XM12O40]n−, where X is the heteroatom, M is the addendum atom, and O represents oxygen. The structure self-assembles in acidic aqueous solution and is a commonly used type of polyoxometalate catalysts.
Zinc compounds are chemical compounds containing the element zinc which is a member of the group 12 of the periodic table. The oxidation state of zinc in most compounds is the group oxidation state of +2. Zinc may be classified as a post-transition main group element with zinc(II). Zinc compounds are noteworthy for their nondescript appearance and behavior: they are generally colorless, do not readily engage in redox reactions, and generally adopt symmetrical structures.
In chemistry, ion association is a chemical reaction whereby ions of opposite electric charge come together in solution to form a distinct chemical entity. Ion associates are classified, according to the number of ions that associate with each other, as ion pairs, ion triplets, etc. Ion pairs are also classified according to the nature of the interaction as contact, solvent-shared or solvent-separated. The most important factor to determine the extent of ion association is the dielectric constant of the solvent. Ion associates have been characterized by means of vibrational spectroscopy, as introduced by Niels Bjerrum, and dielectric-loss spectroscopy.
In chemistry, metal aquo complexes are coordination compounds containing metal ions with only water as a ligand. These complexes are the predominant species in aqueous solutions of many metal salts, such as metal nitrates, sulfates, and perchlorates. They have the general stoichiometry [M(H2O)n]z+. Their behavior underpins many aspects of environmental, biological, and industrial chemistry. This article focuses on complexes where water is the only ligand, but of course many complexes are known to consist of a mix of aquo and other ligands.
The hydration number of a compound is defined as the number of molecules of water bonded to a central ion, often a metal cation. The hydration number is related to the broader concept of solvation number, the number of solvent molecules bonded to a central atom. The hydration number varies with the atom or ion of interest.
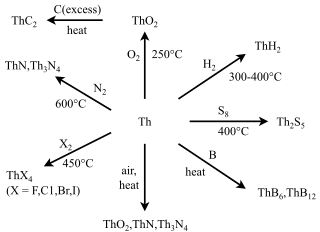
Many compounds of thorium are known: this is because thorium and uranium are the most stable and accessible actinides and are the only actinides that can be studied safely and legally in bulk in a normal laboratory. As such, they have the best-known chemistry of the actinides, along with that of plutonium, as the self-heating and radiation from them is not enough to cause radiolysis of chemical bonds as it is for the other actinides. While the later actinides from americium onwards are predominantly trivalent and behave more similarly to the corresponding lanthanides, as one would expect from periodic trends, the early actinides up to plutonium have relativistically destabilised and hence delocalised 5f and 6d electrons that participate in chemistry in a similar way to the early transition metals of group 3 through 8: thus, all their valence electrons can participate in chemical reactions, although this is not common for neptunium and plutonium.
Aluminium (British and IUPAC spellings) or aluminum (North American spelling) combines characteristics of pre- and post-transition metals. Since it has few available electrons for metallic bonding, like its heavier group 13 congeners, it has the characteristic physical properties of a post-transition metal, with longer-than-expected interatomic distances. Furthermore, as Al3+ is a small and highly charged cation, it is strongly polarizing and aluminium compounds tend towards covalency; this behaviour is similar to that of beryllium (Be2+), an example of a diagonal relationship. However, unlike all other post-transition metals, the underlying core under aluminium's valence shell is that of the preceding noble gas, whereas for gallium and indium it is that of the preceding noble gas plus a filled d-subshell, and for thallium and nihonium it is that of the preceding noble gas plus filled d- and f-subshells. Hence, aluminium does not suffer the effects of incomplete shielding of valence electrons by inner electrons from the nucleus that its heavier congeners do. Aluminium's electropositive behavior, high affinity for oxygen, and highly negative standard electrode potential are all more similar to those of scandium, yttrium, lanthanum, and actinium, which have ds2 configurations of three valence electrons outside a noble gas core: aluminium is the most electropositive metal in its group. Aluminium also bears minor similarities to the metalloid boron in the same group; AlX3 compounds are valence isoelectronic to BX3 compounds (they have the same valence electronic structure), and both behave as Lewis acids and readily form adducts. Additionally, one of the main motifs of boron chemistry is regular icosahedral structures, and aluminium forms an important part of many icosahedral quasicrystal alloys, including the Al–Zn–Mg class.
Cobalt compounds are chemical compounds formed by cobalt with other elements.
↑ Johansson, Georg (1992). Sykes, A.G. (ed.). Structures of Compexes in Solution Derived from X-ray Diffraction Measurements. Advances in Inorganic Chemistry. Vol.39. San Diego: Academic Press. pp.161–232. ISBN978-0-12-023639-8.
↑ Ohtaki, H.; Radnai, T. (1993). "Structure and dynamics of hydrated ions". Chem. Rev. 93 (3): 1157–1204. doi:10.1021/cr00019a014.
↑ Magini, M.; Licheri, G.; Paschina, G.; Piccaluga,G.; Pinna, G. (1988). X-ray diffraction of ions in aqueous solutions: hydration and complex formation. Boca Raton, Fla: CRC Press. ISBN978-0-8493-6945-2.
↑ Enderby, J.E.; Nielson, G.W. (1989). Sykes, A.G. (ed.). The Coordination of Metal Aquaions. Advances in Inorganic Chemistry. Vol.34. San Diego: Academic Press. pp.195–218. doi:10.1016/S0898-8838(08)60017-3. ISBN978-0-12-023634-3.
↑ Enderby, J.E. (1987). "Diffraction Studies of Aqueous Ionic Solutions". In Bellisent-Funel, M-C.; Neilson, G.W. (eds.). The Physics and Chemistry of Aqueous Solutions. NATO ASI Series. Reidel. pp.129–145. ISBN978-90-277-2534-9. p. 138.
↑ Frank, Patrick; Benfatto, Maurizio; Szilagyi, Robert K.; D'Angelo, Paola; Della Longa, Stefano; Hodgson, Keith O. (2005). "The Solution Structure of [Cu(aq)]2+ and Its Implications for Rack-Induced Bonding in Blue Copper Protein Active Sites". Inorganic Chemistry. 44 (6): 1922–1933. doi:10.1021/ic0400639. PMID15762718.
↑ Finholt, James E.; Leupin, Peter; Sykes, A. Geoffrey (1983). "Kinetics and mechanism of substitution of the quadruply bonded molybdenum(II) aqua dimer with thiocyanate and oxalate". Inorganic Chemistry. 22 (22): 3315–3333. doi:10.1021/ic00164a027.
↑ Pan, Kuan; Fu, Yi-Chang; Huang, Teh-Shoon (December 1964). "Polarographic Behavior of Germanium(II)‐Perchlorate in Perchloric Acid Solutions". Journal of the Chinese Chemical Society. 11 (4): 176–184. doi:10.1002/jccs.196400020.
↑ Azam, S. S.; Lim, L.; Hofer, T. S.; Randolf, B. R.; Rode, B. M. (2009). "Hydrated Germanium (II): Irregular Structural and Dynamical Properties Revealed by a Quantum Mechanical Charge Field Molecular Dynamics Study". Journal of Computational Chemistry. 31 (2): 278–285. doi:10.1002/jcc.21315. PMID19479764. S2CID22766649.
↑ Persson, Ingmar; Lyczko, Krzysztof; Lundberg, Daniel; Eriksson, Lars; Płaczek, Anna (2011). "Coordination Chemistry Study of Hydrated and Solvated Lead(II) Ions in Solution and Solid State". Inorganic Chemistry. 50 (3): 1058–1072. doi:10.1021/ic1017714. PMID21226482.
↑ Bhattacharjee, Anirban; Hofer, Thomas S.; Pribil, Andreas B.; Randolf, Bernhard R.; Rode, Bernd M. (2009). "Hydrolysis of As(III): A femtosecond process". Chemical Physics Letters. 473 (1–3): 176–178. Bibcode:2009CPL...473..176B. doi:10.1016/j.cplett.2009.03.011.
↑ Jander, G.; Hartmann, H.-J. (1965). "Über Reaktionen von Antimon(III) in wäßriger Lösung. III: Polarographische Messungen". Zeitschrift für anorganische und allgemeine Chemie (in German). 339 (5–6): 256–261. doi:10.1002/zaac.19653390505.
↑ Lim, Len Herald V.; Bhattacharjee, Anirban; Asam, S. Sikander; Hofer, Thomas S.; Randolf, Bernhard R.; Rode, Bernd M. (2010). "Structural and Dynamical Aspects of the Unsymmetric Hydration of Sb(III): An ab initio Quantum Mechanical Charge Field Molecular Dynamics Simulation". Inorganic Chemistry. 49 (5): 2132–2140. doi:10.1021/ic901737y. PMID20121188.
↑ Ayala, Regla; Martínez, José Manuel; Pappalardo, Rafael R.; Sánchez Marcos, Enrique (2012). "Quantum-Mechanical Study on the Aquaions and Hydrolyzed Species of Po(IV), Te(IV), and Bi(III) in Water". The Journal of Physical Chemistry B. 116 (51): 14903–14914. doi:10.1021/jp309439f.
↑ Robertson, William H.; Johnson, Mark A. (2003). "Molecular Aspects of Halide Ion Hydration: The Cluster Approach". Annual Review of Physical Chemistry. 54 (1): 173–213. doi:10.1146/annurev.physchem.54.011002.103801.
↑ Kugler, H. K.; Keller, C. (1985). 'At, Astatine', System No. 8a. Gmelin Handbook of Inorganic and Organometallic Chemistry. Vol.8 (8thed.). Springer-Verlag. pp.220–221. ISBN978-3-540-93516-2.
Baes, C.F.; Mesmer, R.E. (1986) [1976]. The Hydrolysis of Cations. Malabar, FL: Robert E. Krieger. ISBN978-0-89874-892-5.
Burgess, John (1978). Metal Ions in Solution. Chichester: Ellis Horwood. ISBN978-0-85312-027-8.
Richens, David. T. (1997). The Chemistry of Aqua Ions. Wiley. ISBN978-0-471-97058-3.
Stumm, Werner; Morgan, James J. (1995). Aquatic Chemistry - Chemical Equilibria and Rates in Natural Waters (3rd.ed.). Wiley-Blackwell. ISBN978-0-471-51185-4.
Schweitzer, George K.; Pesterfield, Lester L. (2010). The Aqueous Chemistry of the Elements. Oxford: OUP. ISBN978-0-19-539335-4.
Further reading
H. L. Friedman, F. Franks, Aqueous Solutions of Simple Electrolytes, Springer; reprint of the 1973 edition, 2012 ISBN1468429574
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.
![Schematic representation of the aqua ion [Na(H2O)6] . The oxygen atoms are arranged at the vertices of a regular octahedron centered on the sodium ion. Na+H2O.svg](http://upload.wikimedia.org/wikipedia/commons/thumb/6/67/Na%2BH2O.svg/220px-Na%2BH2O.svg.png)




![Jahn-Teller distorted octahedral structure of [Cu(H2O)6] found in the solid state and possibly also in solution Hexaaquacopper(II)-3D-balls.png](http://upload.wikimedia.org/wikipedia/commons/thumb/0/09/Hexaaquacopper%28II%29-3D-balls.png/180px-Hexaaquacopper%28II%29-3D-balls.png)
![Proposed square pyramidal structure of [Cu(H2O)5] in aqueous solution Cu water.png](http://upload.wikimedia.org/wikipedia/commons/thumb/a/a7/Cu_water.png/180px-Cu_water.png)











![The molybdenum(IV) oxo-aqua ion [Mo3O4(H2O)9] Mo3O4.png](http://upload.wikimedia.org/wikipedia/commons/6/6f/Mo3O4.png)