In a chemical reaction, chemical equilibrium is the state in which both the reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system. This state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but they are equal. Thus, there are no net changes in the concentrations of the reactants and products. Such a state is known as dynamic equilibrium.

Osmotic pressure is the minimum pressure which needs to be applied to a solution to prevent the inward flow of its pure solvent across a semipermeable membrane. It is also defined as the measure of the tendency of a solution to take in its pure solvent by osmosis. Potential osmotic pressure is the maximum osmotic pressure that could develop in a solution if it were separated from its pure solvent by a semipermeable membrane
Raoult's law ( law) is a relation of physical chemistry, with implications in thermodynamics. Proposed by French chemist François-Marie Raoult in 1887, it states that the partial pressure of each component of an ideal mixture of liquids is equal to the vapor pressure of the pure component multiplied by its mole fraction in the mixture. In consequence, the relative lowering of vapor pressure of a dilute solution of nonvolatile solute is equal to the mole fraction of solute in the solution.
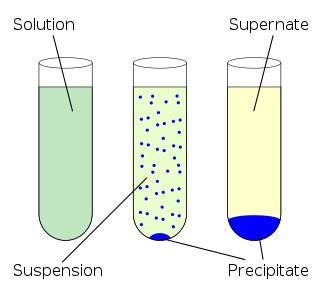
In chemistry, solubility is the ability of a substance, the solute, to form a solution with another substance, the solvent. Insolubility is the opposite property, the inability of the solute to form such a solution.
Solubility equilibrium is a type of dynamic equilibrium that exists when a chemical compound in the solid state is in chemical equilibrium with a solution of that compound. The solid may dissolve unchanged, with dissociation, or with chemical reaction with another constituent of the solution, such as acid or alkali. Each solubility equilibrium is characterized by a temperature-dependent solubility product which functions like an equilibrium constant. Solubility equilibria are important in pharmaceutical, environmental and many other scenarios.
In electrochemistry, the Nernst equation is a chemical thermodynamical relationship that permits the calculation of the reduction potential of a reaction from the standard electrode potential, absolute temperature, the number of electrons involved in the redox reaction, and activities of the chemical species undergoing reduction and oxidation respectively. It was named after Walther Nernst, a German physical chemist who formulated the equation.
In physical chemistry, Henry's law is a gas law that states that the amount of dissolved gas in a liquid is directly proportional to its partial pressure above the liquid. The proportionality factor is called Henry's law constant. It was formulated by the English chemist William Henry, who studied the topic in the early 19th century. In simple words, we can say that the partial pressure of a gas in vapour phase is directly proportional to the mole fraction of a gas in solution.
The self-ionization of water (also autoionization of water, and autodissociation of water, or simply dissociation of water) is an ionization reaction in pure water or in an aqueous solution, in which a water molecule, H2O, deprotonates (loses the nucleus of one of its hydrogen atoms) to become a hydroxide ion, OH−. The hydrogen nucleus, H+, immediately protonates another water molecule to form a hydronium cation, H3O+. It is an example of autoprotolysis, and exemplifies the amphoteric nature of water.
In chemistry, colligative properties are those properties of solutions that depend on the ratio of the number of solute particles to the number of solvent particles in a solution, and not on the nature of the chemical species present. The number ratio can be related to the various units for concentration of a solution such as molarity, molality, normality (chemistry), etc. The assumption that solution properties are independent of nature of solute particles is exact only for ideal solutions, which are solutions that exhibit thermodynamic properties analogous to those of an ideal gas, and is approximate for dilute real solutions. In other words, colligative properties are a set of solution properties that can be reasonably approximated by the assumption that the solution is ideal.
In electrochemistry, the standard hydrogen electrode, is a redox electrode which forms the basis of the thermodynamic scale of oxidation-reduction potentials. Its absolute electrode potential is estimated to be 4.44 ± 0.02 V at 25 °C, but to form a basis for comparison with all other electrochemical reactions, hydrogen's standard electrode potential is declared to be zero volts at any temperature. Potentials of all other electrodes are compared with that of the standard hydrogen electrode at the same temperature.
In chemistry, an ideal solution or ideal mixture is a solution that exhibits thermodynamic properties analogous to those of a mixture of ideal gases. The enthalpy of mixing is zero as is the volume change on mixing by definition; the closer to zero the enthalpy of mixing is, the more "ideal" the behavior of the solution becomes. The vapor pressures of the solvent and solute obey Raoult's law and Henry's law, respectively, and the activity coefficient is equal to one for each component.
The equilibrium constant of a chemical reaction is the value of its reaction quotient at chemical equilibrium, a state approached by a dynamic chemical system after sufficient time has elapsed at which its composition has no measurable tendency towards further change. For a given set of reaction conditions, the equilibrium constant is independent of the initial analytical concentrations of the reactant and product species in the mixture. Thus, given the initial composition of a system, known equilibrium constant values can be used to determine the composition of the system at equilibrium. However, reaction parameters like temperature, solvent, and ionic strength may all influence the value of the equilibrium constant.
In chemical thermodynamics, the fugacity of a real gas is an effective partial pressure which replaces the mechanical partial pressure in an accurate computation of chemical equilibrium. It is equal to the pressure of an ideal gas which has the same temperature and molar Gibbs free energy as the real gas.
In thermodynamics, an activity coefficient is a factor used to account for deviation of a mixture of chemical substances from ideal behaviour. In an ideal mixture, the microscopic interactions between each pair of chemical species are the same and, as a result, properties of the mixtures can be expressed directly in terms of simple concentrations or partial pressures of the substances present e.g. Raoult's law. Deviations from ideality are accommodated by modifying the concentration by an activity coefficient. Analogously, expressions involving gases can be adjusted for non-ideality by scaling partial pressures by a fugacity coefficient.
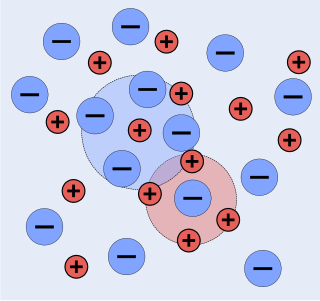
The chemists Peter Debye and Erich Hückel noticed that solutions that contain ionic solutes do not behave ideally even at very low concentrations. So, while the concentration of the solutes is fundamental to the calculation of the dynamics of a solution, they theorized that an extra factor that they termed gamma is necessary to the calculation of the activities of the solution. Hence they developed the Debye–Hückel equation and Debye–Hückel limiting law. The activity is only proportional to the concentration and is altered by a factor known as the activity coefficient . This factor takes into account the interaction energy of ions in solution.

In electrochemistry, and more generally in solution chemistry, a Pourbaix diagram, also known as a potential/pH diagram, EH–pH diagram or a pE/pH diagram, is a plot of possible thermodynamically stable phases of an aqueous electrochemical system. Boundaries (50 %/50 %) between the predominant chemical species are represented by lines. As such a Pourbaix diagram can be read much like a standard phase diagram with a different set of axes. Similarly to phase diagrams, they do not allow for reaction rate or kinetic effects. Beside potential and pH, the equilibrium concentrations are also dependent upon, e.g., temperature, pressure, and concentration. Pourbaix diagrams are commonly given at room temperature, atmospheric pressure, and molar concentrations of 10−6 and changing any of these parameters will yield a different diagram.
An osmotic coefficient is a quantity which characterises the deviation of a solvent from ideal behaviour, referenced to Raoult's law. It can be also applied to solutes. Its definition depends on the ways of expressing chemical composition of mixtures.
In theoretical chemistry, Specific ion Interaction Theory is a theory used to estimate single-ion activity coefficients in electrolyte solutions at relatively high concentrations. It does so by taking into consideration interaction coefficients between the various ions present in solution. Interaction coefficients are determined from equilibrium constant values obtained with solutions at various ionic strengths. The determination of SIT interaction coefficients also yields the value of the equilibrium constant at infinite dilution.
Pitzer equations are important for the understanding of the behaviour of ions dissolved in natural waters such as rivers, lakes and sea-water. They were first described by physical chemist Kenneth Pitzer. The parameters of the Pitzer equations are linear combinations of parameters, of a virial expansion of the excess Gibbs free energy, which characterise interactions amongst ions and solvent. The derivation is thermodynamically rigorous at a given level of expansion. The parameters may be derived from various experimental data such as the osmotic coefficient, mixed ion activity coefficients, and salt solubility. They can be used to calculate mixed ion activity coefficients and water activities in solutions of high ionic strength for which the Debye–Hückel theory is no longer adequate. They are more rigorous than the equations of specific ion interaction theory, but Pitzer parameters are more difficult to determine experimentally than SIT parameters.
Equilibrium chemistry is concerned with systems in chemical equilibrium. The unifying principle is that the free energy of a system at equilibrium is the minimum possible, so that the slope of the free energy with respect to the reaction coordinate is zero. This principle, applied to mixtures at equilibrium provides a definition of an equilibrium constant. Applications include acid–base, host–guest, metal–complex, solubility, partition, chromatography and redox equilibria.











