
The boiling point of a substance is the temperature at which the vapor pressure of a liquid equals the pressure surrounding the liquid and the liquid changes into a vapor.
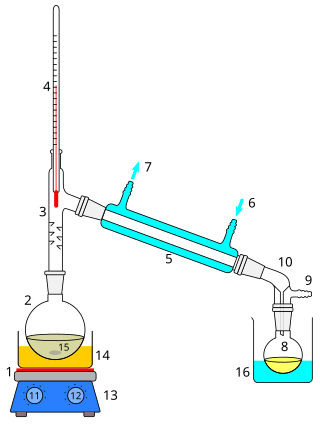
Distillation, also classical distillation, is the process of separating the component substances of a liquid mixture of two or more chemically discrete substances; the separation process is realized by way of the selective boiling of the mixture and the condensation of the vapors in a still.

In the physical sciences, a phase is a region of material that is chemically uniform, physically distinct, and (often) mechanically separable. In a system consisting of ice and water in a glass jar, the ice cubes are one phase, the water is a second phase, and the humid air is a third phase over the ice and water. The glass of the jar is another separate phase.

In chemistry, a solution is a special type of homogeneous mixture composed of two or more substances. In such a mixture, a solute is a substance dissolved in another substance, known as a solvent. If the attractive forces between the solvent and solute particles are greater than the attractive forces holding the solute particles together, the solvent particles pull the solute particles apart and surround them. These surrounded solute particles then move away from the solid solute and out into the solution. The mixing process of a solution happens at a scale where the effects of chemical polarity are involved, resulting in interactions that are specific to solvation. The solution usually has the state of the solvent when the solvent is the larger fraction of the mixture, as is commonly the case. One important parameter of a solution is the concentration, which is a measure of the amount of solute in a given amount of solution or solvent. The term "aqueous solution" is used when one of the solvents is water.
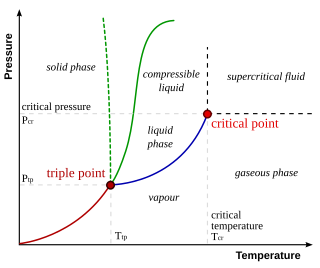
In thermodynamics, the triple point of a substance is the temperature and pressure at which the three phases of that substance coexist in thermodynamic equilibrium. It is that temperature and pressure at which the sublimation, fusion, and vaporisation curves meet. For example, the triple point of mercury occurs at a temperature of −38.8 °C (−37.8 °F) and a pressure of 0.165 mPa.

In physics, a vapor or vapour is a substance in the gas phase at a temperature lower than its critical temperature, which means that the vapor can be condensed to a liquid by increasing the pressure on it without reducing the temperature of the vapor. A vapor is different from an aerosol. An aerosol is a suspension of tiny particles of liquid, solid, or both within a gas.
Raoult's law ( law) is a relation of physical chemistry, with implications in thermodynamics. Proposed by French chemist François-Marie Raoult in 1887, it states that the partial pressure of each component of an ideal mixture of liquids is equal to the vapor pressure of the pure component multiplied by its mole fraction in the mixture. In consequence, the relative lowering of vapor pressure of a dilute solution of nonvolatile solute is equal to the mole fraction of solute in the solution.

Vapor pressure or equilibrium vapor pressure is the pressure exerted by a vapor in thermodynamic equilibrium with its condensed phases at a given temperature in a closed system. The equilibrium vapor pressure is an indication of a liquid's thermodynamic tendency to evaporate. It relates to the balance of particles escaping from the liquid in equilibrium with those in a coexisting vapor phase. A substance with a high vapor pressure at normal temperatures is often referred to as volatile. The pressure exhibited by vapor present above a liquid surface is known as vapor pressure. As the temperature of a liquid increases, the attractive interactions between liquid molecules become less significant in comparison to the entropy of those molecules in the gas phase, increasing the vapor pressure. Thus, liquids with strong intermolecular interactions are likely to have smaller vapor pressures, with the reverse true for weaker interactions.

In a mixture of gases, each constituent gas has a partial pressure which is the notional pressure of that constituent gas as if it alone occupied the entire volume of the original mixture at the same temperature. The total pressure of an ideal gas mixture is the sum of the partial pressures of the gases in the mixture.

A phase diagram in physical chemistry, engineering, mineralogy, and materials science is a type of chart used to show conditions at which thermodynamically distinct phases occur and coexist at equilibrium.
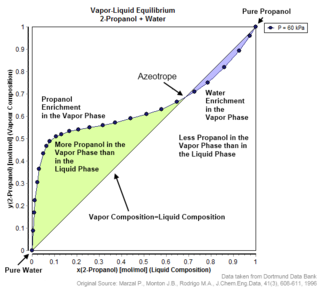
An azeotrope or a constant heating point mixture is a mixture of two or more components in fluidic states whose proportions cannot be altered or changed by simple distillation. This happens because when an azeotrope is boiled, the vapour has the same proportions of constituents as the unboiled mixture. Azeotropic mixture behavior is important for fluid separation processes.
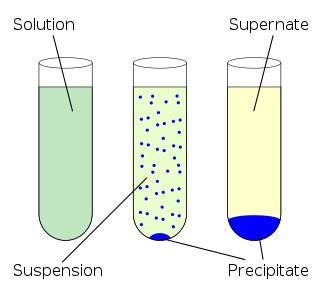
In chemistry, solubility is the ability of a substance, the solute, to form a solution with another substance, the solvent. Insolubility is the opposite property, the inability of the solute to form such a solution.
In chemical thermodynamics, activity is a measure of the "effective concentration" of a species in a mixture, in the sense that the species' chemical potential depends on the activity of a real solution in the same way that it would depend on concentration for an ideal solution. The term "activity" in this sense was coined by the American chemist Gilbert N. Lewis in 1907.
In physical chemistry, Henry's law is a gas law that states that the amount of dissolved gas in a liquid is directly proportional to its partial pressure above the liquid. The proportionality factor is called Henry's law constant. It was formulated by the English chemist William Henry, who studied the topic in the early 19th century. In simple words, we can say that the partial pressure of a gas in vapour phase is directly proportional to the mole fraction of a gas in solution.
In chemistry, colligative properties are those properties of solutions that depend on the ratio of the number of solute particles to the number of solvent particles in a solution, and not on the nature of the chemical species present. The number ratio can be related to the various units for concentration of a solution such as molarity, molality, normality (chemistry), etc. The assumption that solution properties are independent of nature of solute particles is exact only for ideal solutions, which are solutions that exhibit thermodynamic properties analogous to those of an ideal gas, and is approximate for dilute real solutions. In other words, colligative properties are a set of solution properties that can be reasonably approximated by the assumption that the solution is ideal.
In chemical thermodynamics, the fugacity of a real gas is an effective partial pressure which replaces the mechanical partial pressure in an accurate computation of chemical equilibrium. It is equal to the pressure of an ideal gas which has the same temperature and molar Gibbs free energy as the real gas.
In thermodynamics, the entropy of mixing is the increase in the total entropy when several initially separate systems of different composition, each in a thermodynamic state of internal equilibrium, are mixed without chemical reaction by the thermodynamic operation of removal of impermeable partition(s) between them, followed by a time for establishment of a new thermodynamic state of internal equilibrium in the new unpartitioned closed system.
In thermodynamics and chemical engineering, the vapor–liquid equilibrium (VLE) describes the distribution of a chemical species between the vapor phase and a liquid phase.
This glossary of chemistry terms is a list of terms and definitions relevant to chemistry, including chemical laws, diagrams and formulae, laboratory tools, glassware, and equipment. Chemistry is a physical science concerned with the composition, structure, and properties of matter, as well as the changes it undergoes during chemical reactions; it features an extensive vocabulary and a significant amount of jargon.
The Duhem–Margules equation, named for Pierre Duhem and Max Margules, is a thermodynamic statement of the relationship between the two components of a single liquid where the vapour mixture is regarded as an ideal gas:





