
A granuloma is an aggregation of macrophages that forms in response to chronic inflammation. This occurs when the immune system attempts to isolate foreign substances that it is otherwise unable to eliminate. Such substances include infectious organisms including bacteria and fungi, as well as other materials such as foreign objects, keratin, and suture fragments.

Vasculitis is a group of disorders that destroy blood vessels by inflammation. Both arteries and veins are affected. Lymphangitis is sometimes considered a type of vasculitis. Vasculitis is primarily caused by leukocyte migration and resultant damage. Although both occur in vasculitis, inflammation of veins (phlebitis) or arteries (arteritis) on their own are separate entities.

Granulomatosis with polyangiitis (GPA), also known as Wegener's granulomatosis (WG), after the Nazi German physician Friedrich Wegener, is a rare long-term systemic disorder that involves the formation of granulomas and inflammation of blood vessels (vasculitis). It is an autoimmune disease and a form of vasculitis that affects small- and medium-size vessels in many organs but most commonly affects the upper respiratory tract, lungs and kidneys. The signs and symptoms of GPA are highly varied and reflect which organs are supplied by the affected blood vessels. Typical signs and symptoms include nosebleeds, stuffy nose and crustiness of nasal secretions, and inflammation of the uveal layer of the eye. Damage to the heart, lungs and kidneys can be fatal.
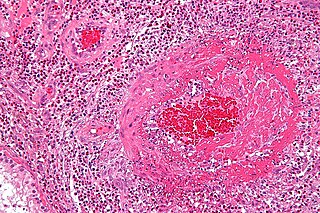
Eosinophilic granulomatosis with polyangiitis (EGPA), formerly known as allergic granulomatosis, is an extremely rare autoimmune condition that causes inflammation of small and medium-sized blood vessels (vasculitis) in persons with a history of airway allergic hypersensitivity (atopy).

Polyarteritis nodosa (PAN) is a systemic necrotizing inflammation of blood vessels (vasculitis) affecting medium-sized muscular arteries, typically involving the arteries of the kidneys and other internal organs but generally sparing the lungs' circulation. Small aneurysms are strung like the beads of a rosary, therefore making this "rosary sign" an important diagnostic feature of the vasculitis. PAN is sometimes associated with infection by the hepatitis B or hepatitis C virus. The condition may be present in infants.
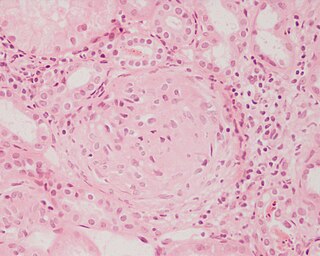
Glomerulonephritis (GN) is a term used to refer to several kidney diseases. Many of the diseases are characterised by inflammation either of the glomeruli or of the small blood vessels in the kidneys, hence the name, but not all diseases necessarily have an inflammatory component.

Anti-neutrophil cytoplasmic antibodies (ANCAs) are a group of autoantibodies, mainly of the IgG type, against antigens in the cytoplasm of neutrophils and monocytes. They are detected as a blood test in a number of autoimmune disorders, but are particularly associated with systemic vasculitis, so called ANCA-associated vasculitides (AAV).

Cryoglobulinemia is a medical condition in which the blood contains large amounts of pathological cold sensitive antibodies called cryoglobulins – proteins that become insoluble at reduced temperatures. This should be contrasted with cold agglutinins, which cause agglutination of red blood cells.

Nephritic syndrome is a syndrome comprising signs of nephritis, which is kidney disease involving inflammation. It often occurs in the glomerulus, where it is called glomerulonephritis. Glomerulonephritis is characterized by inflammation and thinning of the glomerular basement membrane and the occurrence of small pores in the podocytes of the glomerulus. These pores become large enough to permit both proteins and red blood cells to pass into the urine. By contrast, nephrotic syndrome is characterized by proteinuria and a constellation of other symptoms that specifically do not include hematuria. Nephritic syndrome, like nephrotic syndrome, may involve low level of albumin in the blood due to the protein albumin moving from the blood to the urine.
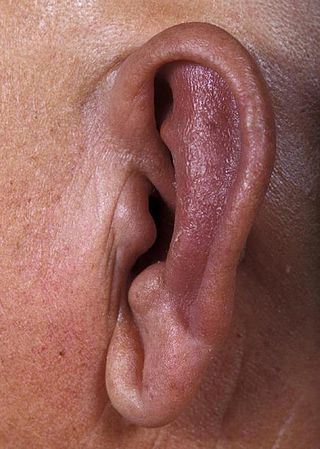
Relapsing polychondritis is a multi-systemic condition characterized by repeated episodes of inflammation and deterioration of cartilage. The often painful disease can cause joint deformity and be life-threatening if the respiratory tract, heart valves, or blood vessels are affected. The exact mechanism is poorly understood, but it is thought to be related to an immune-mediated attack on particular proteins that are abundant in cartilage.
p-ANCA, or MPO-ANCA, or perinuclear anti-neutrophil cytoplasmic antibodies, are antibodies that stain the material around the nucleus of a neutrophil. They are a special class of anti-neutrophil cytoplasmic antibodies.

Rapidly progressive glomerulonephritis (RPGN) is a syndrome of the kidney that is characterized by a rapid loss of kidney function, with glomerular crescent formation seen in at least 50% or 75% of glomeruli seen on kidney biopsies. If left untreated, it rapidly progresses into acute kidney failure and death within months. In 50% of cases, RPGN is associated with an underlying disease such as Goodpasture syndrome, systemic lupus erythematosus or granulomatosis with polyangiitis; the remaining cases are idiopathic. Regardless of the underlying cause, RPGN involves severe injury to the kidneys' glomeruli, with many of the glomeruli containing characteristic glomerular crescents.
Pulmonary-renal syndrome (PRS) is a rare medical syndrome in which respiratory failure involving bleeding in the lungs and kidney failure (glomerulonephritis) occur. PRS is associated with a high rate of morbidity and death. The term was first used by Goodpasture in 1919 to describe the association of respiratory and kidney failure.
Pauci-immune vasculitis is a form of vasculitis that is associated with minimal evidence of hypersensitivity upon immunofluorescent staining for IgG. Often, this is discovered in the setting of the kidney.
Necrotizing vasculitis, also called systemic necrotizing vasculitus, is a category of vasculitis, comprising vasculitides that present with necrosis. Examples include giant cell arteritis, microscopic polyangiitis, and granulomatosis with polyangiitis. ICD-10 uses the variant "necrotizing vasculopathy". ICD-9, while classifying these conditions together, does not use a dedicated phrase, instead calling them "polyarteritis nodosa and allied conditions".

Cutaneous small-vessel vasculitis (CSVV), is inflammation of small blood vessels, usually accompanied by small lumps beneath the skin. The condition is also known as hypersensitivity vasculitis, cutaneous leukocytoclastic vasculitis, hypersensitivity angiitis, cutaneous leukocytoclastic angiitis, cutaneous necrotizing vasculitis and cutaneous necrotizing venulitis,

Retinal vasculitis is inflammation of the vascular branches of the retinal artery, caused either by primary ocular disease processes, or as a specific presentation of any systemic form of vasculitis such as Behçet's disease, sarcoidosis, multiple sclerosis, or any form of systemic necrotizing vasculitis such as temporal arteritis, polyarteritis nodosa, and granulomatosis with polyangiitis, or due to lupus erythematosus, or rheumatoid arthritis. Eales disease, pars planitis, birdshot retinochoroidopathy, and Fuchs heterochromic iridocyclitis (FHI) can also cause retinal vasculitis. Infectious pathogens such as Mycobacterium tuberculosis, visceral larva migrans can also cause retinal vasculitis. Drug-induced vasculitis may involve retina as well, as seen in methamphetamine induced vasculitis.
Ronald Jonathan Falk, MD, FACP, FASN is the Nan and Hugh Cullman Eminent Professor and Chair of the Department of Medicine at the University of North Carolina-Chapel Hill (UNC). He is a clinical nephrologist and internationally recognized expert in anti-neutrophil cytoplasmic autoantibody (ANCA)-induced vasculitis and autoimmune kidney disease. His career as a translational physician-scientist spans more than three decades. His clinical practice and translational research focus on characterizing the cell, tissue and physiologic changes in the development of specific autoimmune kidney diseases and developing new approaches for studying autoimmunity, inflammation and basic neutrophil/monocyte biology. He was Chief of the UNC Division of Nephrology and Hypertension from 1993-2015. He co-founded the UNC Kidney Center in 2005 and continues as Co-Director. Falk is a Past-President of the American Society of Nephrology (ASN). Since 2015, he has served as Chair of the Department of Medicine at UNC.
Vasculitic neuropathy is a peripheral neuropathic disease. In a vasculitic neuropathy there is damage to the vessels that supply blood to the nerves. It can be as part of a systemic problem or can exist as a single-organ issue only affecting the peripheral nervous system (PNS). It is diagnosed with the use of electrophysiological testing, blood tests, nerve biopsy and clinical examination. It is a serious medical condition that can cause prolonged morbidity and disability and generally requires treatment. Treatment depends on the type but it is mostly with corticosteroids or immunomodulating therapies.

Avacopan, sold under the brand name Tavneos, is a medication used to treat anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Avacopan is a complement 5a receptor antagonist and a cytochrome P450 3A4 inhibitor.