
Polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation. It is fundamental to many of the procedures used in genetic testing and research, including analysis of ancient samples of DNA and identification of infectious agents. Using PCR, copies of very small amounts of DNA sequences are exponentially amplified in a series of cycles of temperature changes. PCR is now a common and often indispensable technique used in medical laboratory research for a broad variety of applications including biomedical research and criminal forensics.

Reverse transcription polymerase chain reaction (RT-PCR) is a laboratory technique combining reverse transcription of RNA into DNA and amplification of specific DNA targets using polymerase chain reaction (PCR). It is primarily used to measure the amount of a specific RNA. This is achieved by monitoring the amplification reaction using fluorescence, a technique called real-time PCR or quantitative PCR (qPCR). Combined RT-PCR and qPCR are routinely used for analysis of gene expression and quantification of viral RNA in research and clinical settings.
In molecular biology, an amplicon is a piece of DNA or RNA that is the source and/or product of amplification or replication events. It can be formed artificially, using various methods including polymerase chain reactions (PCR) or ligase chain reactions (LCR), or naturally through gene duplication. In this context, amplification refers to the production of one or more copies of a genetic fragment or target sequence, specifically the amplicon. As it refers to the product of an amplification reaction, amplicon is used interchangeably with common laboratory terms, such as "PCR product."

Fluorescence in situ hybridization (FISH) is a molecular cytogenetic technique that uses fluorescent probes that bind to only particular parts of a nucleic acid sequence with a high degree of sequence complementarity. It was developed by biomedical researchers in the early 1980s to detect and localize the presence or absence of specific DNA sequences on chromosomes. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes. FISH is often used for finding specific features in DNA for use in genetic counseling, medicine, and species identification. FISH can also be used to detect and localize specific RNA targets in cells, circulating tumor cells, and tissue samples. In this context, it can help define the spatial-temporal patterns of gene expression within cells and tissues.
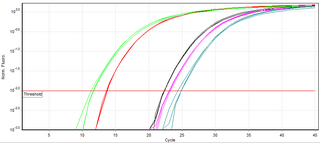
A real-time polymerase chain reaction is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. A SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.

Bisulfitesequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
Digital polymerase chain reaction is a biotechnological refinement of conventional polymerase chain reaction methods that can be used to directly quantify and clonally amplify nucleic acids strands including DNA, cDNA, or RNA. The key difference between dPCR and traditional PCR lies in the method of measuring nucleic acids amounts, with the former being a more precise method than PCR, though also more prone to error in the hands of inexperienced users. A "digital" measurement quantitatively and discretely measures a certain variable, whereas an “analog” measurement extrapolates certain measurements based on measured patterns. PCR carries out one reaction per single sample. dPCR also carries out a single reaction within a sample, however the sample is separated into a large number of partitions and the reaction is carried out in each partition individually. This separation allows a more reliable collection and sensitive measurement of nucleic acid amounts. The method has been demonstrated as useful for studying variations in gene sequences — such as copy number variants and point mutations — and it is routinely used for clonal amplification of samples for next-generation sequencing.
Loop-mediated isothermal amplification (LAMP) is a single-tube technique for the amplification of DNA and a low-cost alternative to detect certain diseases. Reverse transcription loop-mediated isothermal amplification (RT-LAMP) combines LAMP with a reverse transcription step to allow the detection of RNA.

A nucleic acid test (NAT) is a technique used to detect a particular nucleic acid sequence and thus usually to detect and identify a particular species or subspecies of organism, often a virus or bacterium that acts as a pathogen in blood, tissue, urine, etc. NATs differ from other tests in that they detect genetic materials rather than antigens or antibodies. Detection of genetic materials allows an early diagnosis of a disease because the detection of antigens and/or antibodies requires time for them to start appearing in the bloodstream. Since the amount of a certain genetic material is usually very small, many NATs include a step that amplifies the genetic material—that is, makes many copies of it. Such NATs are called nucleic acid amplification tests (NAATs). There are several ways of amplification, including polymerase chain reaction (PCR), strand displacement assay (SDA), or transcription mediated assay (TMA).
The versatility of polymerase chain reaction (PCR) has led to a large number of variants of PCR.
OLIGO Primer Analysis Software was the first publicly available software for DNA primer design. The first papers describing this software were published in 1989 and 1990, and consecutive upgrades in the 1990s and 2000s, all have been cited together over 600 times in scientific journals and over 500 times in patents. The program is a comprehensive real time PCR primer and probe search and analysis tool, and also does other tasks such as siRNA and molecular beacon searches, open reading frame and restriction enzyme analysis etc. It has been created and maintained by Wojciech Rychlik and Piotr Rychlik. The OLIGO has been reviewed several times in scientific journals, for the first time in 1991 in a review in Critical Reviews in Biochemistry and Molecular Biology, and for its next upgrades.
High Resolution Melt (HRM) analysis is a powerful technique in molecular biology for the detection of mutations, polymorphisms and epigenetic differences in double-stranded DNA samples. It was discovered and developed by Idaho Technology and the University of Utah. It has advantages over other genotyping technologies, namely:

Diversity Arrays Technology (DArT) is a high-throughput genetic marker technique that can detect allelic variations to provides comprehensive genome coverage without any DNA sequence information for genotyping and other genetic analysis. The general steps involve reducing the complexity of the genomic DNA with specific restriction enzymes, choosing diverse fragments to serve as representations for the parent genomes, amplify via polymerase chain reaction (PCR), insert fragments into a vector to be placed as probes within a microarray, then fluorescent targets from a reference sequence will be allowed to hybridize with probes and put through an imaging system. The objective is to identify and quantify various forms of DNA polymorphism within genomic DNA of sampled species.
Multiplex polymerase chain reaction refers to the use of polymerase chain reaction to amplify several different DNA sequences simultaneously. This process amplifies DNA in samples using multiple primers and a temperature-mediated DNA polymerase in a thermal cycler. The primer design for all primers pairs has to be optimized so that all primer pairs can work at the same annealing temperature during PCR.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.
Polony sequencing is an inexpensive but highly accurate multiplex sequencing technique that can be used to “read” millions of immobilized DNA sequences in parallel. This technique was first developed by Dr. George Church's group at Harvard Medical School. Unlike other sequencing techniques, Polony sequencing technology is an open platform with freely downloadable, open source software and protocols. Also, the hardware of this technique can be easily set up with a commonly available epifluorescence microscopy and a computer-controlled flowcell/fluidics system. Polony sequencing is generally performed on paired-end tags library that each molecule of DNA template is of 135 bp in length with two 17–18 bp paired genomic tags separated and flanked by common sequences. The current read length of this technique is 26 bases per amplicon and 13 bases per tag, leaving a gap of 4–5 bases in each tag.
9q34 deletion syndrome is a rare genetic disorder. Terminal deletions of chromosome 9q34 have been associated with childhood hypotonia, a distinctive facial appearance and developmental disability. The facial features typically described include arched eyebrows, small head circumference, midface hypoplasia, prominent jaw and a pouting lower lip. Individuals with this disease may often have speech impediments, such as speech delays. Other characteristics of this disease include: epilepsy, congenital and urogenital defects, microcephaly, corpulence, and psychiatric disorders. From analysis of chromosomal breakpoints, as well as gene sequencing in suggestive cases, Kleefstra and colleagues identified EHMT1 as the causative gene. This gene is responsible for producing the protein histone methyltransferase which functions to alter histones. Ultimately, histone methyltransferases are important in deactivating certain genes, needed for proper growth and development. Moreover, a frameshift, missense, or nonsense error in the coding sequence of EHMT1 can result in this condition in an individual.
Recombinase polymerase amplification (RPA) is a single tube, isothermal alternative to the polymerase chain reaction (PCR). By adding a reverse transcriptase enzyme to an RPA reaction it can detect RNA as well as DNA, without the need for a separate step to produce cDNA,. Because it is isothermal, RPA can use much simpler equipment than PCR, which requires a thermal cycler. Operating best at temperatures of 37–42 °C and still working, albeit more slowly, at room temperature means RPA reactions can in theory be run quickly simply by holding a tube. This makes RPA an excellent candidate for developing low-cost, rapid, point-of-care molecular tests. An international quality assessment of molecular detection of Rift Valley fever virus performed as well as the best RT-PCR tests, detecting less concentrated samples missed by some PCR tests and an RT-LAMP test. RPA was developed and launched by TwistDx Ltd., a biotechnology company based in Cambridge, UK.
Johannes Petrus Schouten is a Dutch scientist, entrepreneur and philanthropist. He is the inventor of Multiplex-Ligation-dependent Probe Amplification (MLPA), a method for the research of genetic disorders. He founded the biotech company MRC Holland, based in Amsterdam and the MRC Holland Foundation.

