The urea cycle (also known as the ornithine cycle) is a cycle of biochemical reactions that produces urea (NH2)2CO from ammonia (NH3). Animals that use this cycle, mainly amphibians and mammals, are called ureotelic.

Ornithine transcarbamylase (OTC) is an enzyme that catalyzes the reaction between carbamoyl phosphate (CP) and ornithine (Orn) to form citrulline (Cit) and phosphate (Pi). There are two classes of OTC: anabolic and catabolic. This article focuses on anabolic OTC. Anabolic OTC facilitates the sixth step in the biosynthesis of the amino acid arginine in prokaryotes. In contrast, mammalian OTC plays an essential role in the urea cycle, the purpose of which is to capture toxic ammonia and transform it into urea, a less toxic nitrogen source, for excretion.

Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to brain injury and death. It may be primary or secondary.

Ornithine transcarbamylase deficiency also known as OTC deficiency is the most common urea cycle disorder in humans. Ornithine transcarbamylase, the defective enzyme in this disorder is the final enzyme in the proximal portion of the urea cycle, responsible for converting carbamoyl phosphate and ornithine into citrulline. OTC deficiency is inherited in an X-linked recessive manner, meaning males are more commonly affected than females.

Citrullinemia is an autosomal recessive urea cycle disorder that causes ammonia and other toxic substances to accumulate in the blood.

Carnitine-acylcarnitine translocase deficiency is a rare, autosomal recessive metabolic disorder that prevents the body from converting long-chain fatty acids into energy, particularly during periods without food. Carnitine, a natural substance acquired mostly through the diet, is used by cells to process fats and produce energy. People with this disorder have a faulty enzyme that prevents long-chain fatty acids from being transported into the innermost part of the mitochondria for processing.

Lysinuric protein intolerance (LPI) is an autosomal recessive metabolic disorder affecting amino acid transport.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

Methylmalonyl-CoA mutase is a mitochondrial homodimer apoenzyme that focuses on the catalysis of methylmalonyl CoA to succinyl CoA. The enzyme is bound to adenosylcobalamin, a hormonal derivative of vitamin B12 in order to function. Methylmalonyl-CoA mutase deficiency is caused by genetic defect in the MUT gene responsible for encoding the enzyme. Deficiency in this enzyme accounts for 60% of the cases of methylmalonic acidemia.

Sodium phenylbutyrate, sold under the brand name Buphenyl among others, is a salt of an aromatic fatty acid, 4-phenylbutyrate (4-PBA) or 4-phenylbutyric acid. The compound is used to treat urea cycle disorders, because its metabolites offer an alternative pathway to the urea cycle to allow excretion of excess nitrogen.

N-Acetylglutamate synthase deficiency is an autosomal recessive urea cycle disorder.
Carbamoyl phosphate synthetase I deficiency is an autosomal recessive metabolic disorder that causes ammonia to accumulate in the blood due to a lack of the enzyme carbamoyl phosphate synthetase I. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
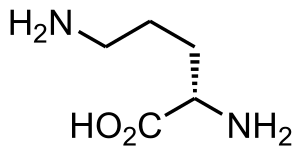
Ornithine translocase is responsible for transporting ornithine from the cytosol into the mitochondria in the urea cycle. It is highly expressed in the liver and pancreas.

Orotic aciduria is a disease caused by an enzyme deficiency, resulting in a decreased ability to synthesize pyrimidines. It was the first described enzyme deficiency of the de novo pyrimidine synthesis pathway.

Argininemia is an autosomal recessive urea cycle disorder where a deficiency of the enzyme arginase causes a buildup of arginine and ammonia in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if levels become too high; the nervous system is especially sensitive to the effects of excess ammonia.
Solute carrier family 25, member 29 is a protein that in humans is encoded by the SLC25A29 gene. The gene is also known as CACL and C14orf69. SLC25A29 belongs to a protein family of solute carriers called the mitochondrial carriers.
Transient hyperammonemia of the newborn (THAN) is an idiopathic disorder occasionally present in preterm newborns but not always symptomatic. Continuous dialysis or hemofiltration have proven to be the most effective treatment. Nutritional support and sodium benzoate have also been used to treat THAN.
Ornithine aminotransferase deficiency is an inborn error of ornithine metabolism, caused by decreased activity of the enzyme ornithine aminotransferase. Biochemically, it can be detected by elevated levels of ornithine in the blood. Clinically, it presents initially with poor night vision, which slowly progresses to total blindness. It is believed to be inherited in an autosomal recessive manner. Approximately 200 known cases have been reported in the literature. The incidence is highest in Finland, estimated at 1:50,000.

L-Homocitrulline is an amino acid and a metabolite of ornithine in mammalian metabolism. The amino acid can be detected in larger amounts in the urine of individuals with urea cycle disorders. At present, it is thought that the depletion of the ornithine supply causes the accumulation of carbamyl-phosphate in the urea cycle which may be responsible for the enhanced synthesis of homocitrulline and homoarginine. Both amino acids can be detected in urine. Amino acid analysis allows for the quantitative analysis of these amino acid metabolites in biological fluids such as urine or blood.

Citrullinemia type I (CTLN1), also known as arginosuccinate synthetase deficiency, is a rare disease caused by a deficiency in argininosuccinate synthetase, an enzyme involved in excreting excess nitrogen from the body. There are mild and severe forms of the disease, which is one of the urea cycle disorders.