
Methylmalonic acidemia, also called methylmalonic aciduria, is an autosomal recessive metabolic disorder that disrupts normal amino acid metabolism. It is a classical type of organic acidemia. The result of this condition is the inability to properly digest specific fats and proteins, which in turn leads to a buildup of a toxic level of methylmalonic acid in the blood.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.

Maple syrup urine disease (MSUD) is an autosomal recessive metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax, particularly prior to diagnosis and during times of acute illness.

Carnitine palmitoyltransferase I deficiency is a rare metabolic disorder that prevents the body from converting certain fats called long-chain fatty acids into energy, particularly during periods without food. It is caused by a mutation in CPT1A on chromosome 11.

Carnitine-acylcarnitine translocase deficiency is a rare, autosomal recessive metabolic disorder that prevents the body from converting long-chain fatty acids into energy, particularly during periods without food. Carnitine, a natural substance acquired mostly through the diet, is used by cells to process fats and produce energy. People with this disorder have a faulty enzyme that prevents long-chain fatty acids from being transported into the innermost part of the mitochondria for processing.
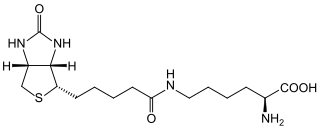
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

Malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA into acetyl-CoA and carbon dioxide.

Glycine encephalopathy is a rare autosomal recessive disorder of glycine metabolism. After phenylketonuria, glycine encephalopathy is the second most common disorder of amino acid metabolism. The disease is caused by defects in the glycine cleavage system, an enzyme responsible for glycine catabolism. There are several forms of the disease, with varying severity of symptoms and time of onset. The symptoms are exclusively neurological in nature, and clinically this disorder is characterized by abnormally high levels of the amino acid glycine in bodily fluids and tissues, especially the cerebrospinal fluid.
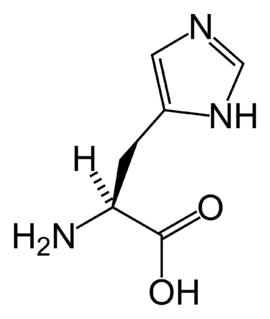
Histidinemia is a rare autosomal recessive metabolic disorder caused by a deficiency of the enzyme histidase. Histidase is needed for the metabolism of the amino acid histidine. Although originally thought to be linked to multiple developmental disorders histidinemia is now accepted as a relatively benign disorder, leading to a reduction in the prevalence of neonatal screening procedures.
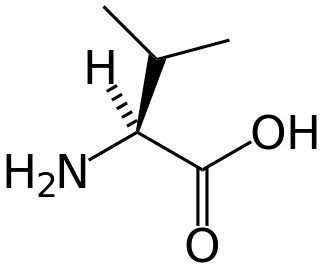
Isobutyryl-coenzyme A dehydrogenase deficiency is a rare metabolic disorder in which the body is unable to process certain amino acids properly.

Urocanic acid is an intermediate in the catabolism of L-histidine.

Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.

Hypermethioninemia is an excess of the amino acid methionine, in the blood. This condition can occur when methionine is not broken down properly in the body.

Hyperprolinemia is a condition which occurs when the amino acid proline is not broken down properly by the enzymes proline oxidase or pyrroline-5-carboxylate dehydrogenase, causing a buildup of proline in the body.
Hypervalinemia is a rare autosomal recessive metabolic disorder in which urinary and serum levels of the branched-chain amino acid valine are elevated, without related elevation of the branched-chain amino acids leucine and isoleucine. It is caused by a deficiency of the enzyme valine transaminase.
Organic acidemia, is a term used to classify a group of metabolic disorders which disrupt normal amino acid metabolism, particularly branched-chain amino acids, causing a buildup of acids which are usually not present.

Carnosinemia is a rare autosomal recessive metabolic disorder caused by a deficiency of carnosinase, a dipeptidase.
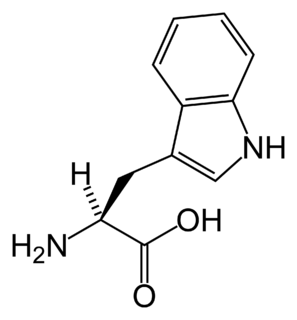
Hypertryptophanemia is a rare autosomal recessive metabolic disorder that results in a massive buildup of the amino acid tryptophan in the blood, with associated symptoms and tryptophanuria.
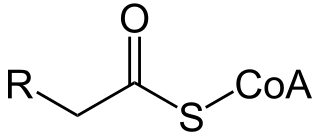
A broad classification for genetic disorders that result from an inability of the body to produce or utilize one enzyme that is required to oxidize fatty acids. The enzyme can be missing or improperly constructed, resulting in it not working. This leaves the body unable to produce energy within the liver and muscles from fatty acid sources.

