
Lichen planus (LP) is a chronic inflammatory and autoimmune disease that affects the skin, nails, hair, and mucous membranes. It is not an actual lichen, but is named for its appearance. It is characterized by polygonal, flat-topped, violaceous papules and plaques with overlying, reticulated, fine white scale, commonly affecting dorsal hands, flexural wrists and forearms, trunk, anterior lower legs and oral mucosa. The hue may be gray-brown in people with darker skin. Although there is a broad clinical range of LP manifestations, the skin and oral cavity remain as the major sites of involvement. The cause is unknown, but it is thought to be the result of an autoimmune process with an unknown initial trigger. There is no cure, but many different medications and procedures have been used in efforts to control the symptoms.
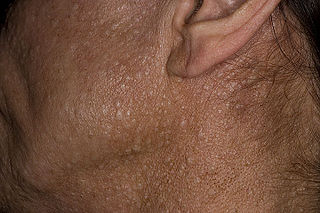
Birt–Hogg–Dubé syndrome (BHD), also Hornstein–Birt–Hogg–Dubé syndrome, Hornstein–Knickenberg syndrome, and fibrofolliculomas with trichodiscomas and acrochordons is a human, adult onset, autosomal dominant genetic disorder caused by a mutation in the folliculin (FLCN) gene. It can cause susceptibility to kidney cancer, renal and pulmonary cysts, and noncancerous tumors of the hair follicles, called fibrofolliculomas. The symptoms seen in each family are unique, and can include any combination of the three symptoms. Fibrofolliculomas are the most common manifestation, found on the face and upper trunk in over 80% of people with BHD over the age of 40. Pulmonary cysts are equally common (84%) and 24% of people with BHD eventually experience a collapsed lung. Kidney tumors, both cancerous and benign, occur in 14–34% of people with BHD; the associated kidney cancers are often rare hybrid tumors.

Neurofibromatosis type II is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves. The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s). Many people with this condition also experience vision problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.
The oral mucosa is the mucous membrane lining the inside of the mouth. It comprises stratified squamous epithelium, termed "oral epithelium", and an underlying connective tissue termed lamina propria. The oral cavity has sometimes been described as a mirror that reflects the health of the individual. Changes indicative of disease are seen as alterations in the oral mucosa lining the mouth, which can reveal systemic conditions, such as diabetes or vitamin deficiency, or the local effects of chronic tobacco or alcohol use. The oral mucosa tends to heal faster and with less scar formation compared to the skin. The underlying mechanism remains unknown, but research suggests that extracellular vesicles might be involved.

A neurofibroma is a benign nerve-sheath tumor in the peripheral nervous system. In 90% of cases, they are found as stand-alone tumors, while the remainder are found in persons with neurofibromatosis type I (NF1), an autosomal-dominant genetically inherited disease. They can result in a range of symptoms from physical disfiguration and pain to cognitive disability.
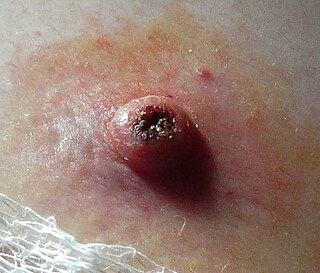
Keratoacanthoma (KA) is a common low-grade rapidly-growing skin tumour that is believed to originate from the hair follicle and can resemble squamous cell carcinoma.
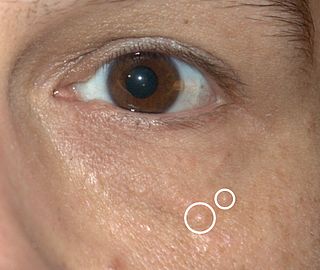
Syringomas are benign eccrine sweat duct tumors, typically found clustered on eyelids, although they may also be found in the armpits, abdomen, chest, neck, scalp, or groin area, including genitals, in a symmetric pattern. They are skin-colored or yellowish firm, rounded bumps, 1–3 mm in diameter, and may be confused with xanthoma, milia, hidrocystoma, trichoepithelioma, and xanthelasma. They are more common in women and are most commonly found in middle-aged Asian women. While they can present at any time in life, they typically present during adolescence. They are usually not associated with any other symptoms, although can sometimes cause itchiness or irritation.
Schwannomatosis is an extremely rare genetic disorder closely related to the more-common disorder neurofibromatosis (NF). Originally described in Japanese patients, it consists of multiple cutaneous schwannomas, central nervous system tumors, and other neurological complications, excluding hallmark signs of NF. The exact frequency of schwannomatosis cases is unknown, although some populations have noted frequencies as few as 1 case per 1.7 million people.

A schwannoma is a usually benign nerve sheath tumor composed of Schwann cells, which normally produce the insulating myelin sheath covering peripheral nerves.

Granular cell tumor is a tumor that can develop on any skin or mucosal surface, but occurs on the tongue 40% of the time.

Juvenile xanthogranuloma is a form of histiocytosis, classified as non-Langerhans cell histiocytosis. It is a rare skin disorder that primarily affects children under one year of age but can also be found in older children and adults.

A Neurothekeoma (NT) is a type of rare benign cutaneous tumor that usually develops on the head and neck. They often occur in the second and early third decades of life and tend to afflict women more frequently than men. First described by Richard L Gallager and Elson B. Helwig, who proposed the term in order to reflect the presumed origin of the lesion from nerve sheath. Microscopically, the lesions described closely resembled the tumor, "nerve sheath myxoma (NSM)", an entity first described by Harkin and Reed. The latter had, through the years, been variously described as bizarre cutaneous neurofibroma, myxoma of nerve sheath, and pacinian neurofibroma.
Neural fibrolipoma is an overgrowth of fibro-fatty tissue along a nerve trunk that often leads to nerve compression. These only occur in the extremities, and often affect the median nerve. They are rare, very slow-growing, and their origin is unknown. It is believed that they may begin growth in response to trauma. They are not encapsulated by any sort of covering or sheath around the growth itself, as opposed to other cysts beneath the skin that often are. This means there are loosely defined margins of this lipoma. Despite this, they are known to be benign. Neural fibrolipomas are often more firm and tough to the touch than other lipomas. They are slightly mobile under the skin, and compress with pressure.
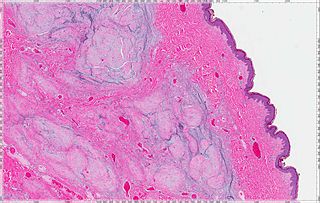
A cutaneous myxoma, or superficial angiomyxoma, consists of a multilobulated myxoid mass containing stellate or spindled fibroblasts with pools of mucin forming cleft-like spaces. There is often a proliferation of blood vessels and an inflammatory infiltrate. Staining is positive for vimentin, negative for cytokeratin and desmin, and variable for CD34, Factor VIIIa, SMA, MSA and S-100.
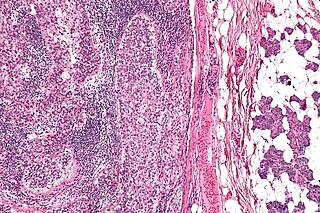
Sebaceous carcinoma, also known as sebaceous gland carcinoma (SGc), sebaceous cell carcinoma, and meibomian gland carcinoma is an uncommon malignant cutaneous tumor. Most are typically about 1.4 cm at presentation. SGc originates from sebaceous glands in the skin and, therefore, may originate anywhere in the body where these glands are found. SGc can be divided into 2 types: periocular and extraocular. The periocular region is rich in sebaceous glands making it a common site of origin. The cause of these lesions in the vast majority of cases is unknown. Occasional cases may be associated with Muir-Torre syndrome. SGc accounts for approximately 0.7% of all skin cancers, and the incidence of SGc is highest in Caucasian, Asian, and Indian populations. Due to the rarity of this tumor and variability in clinical and histological presentation, SGc is often misdiagnosed as an inflammatory condition or a more common neoplasm. SGc is commonly treated with wide local excision or Mohs micrographic surgery, and the relative survival rates at 5 and 10 years are 92.72 and 86.98%, respectively.
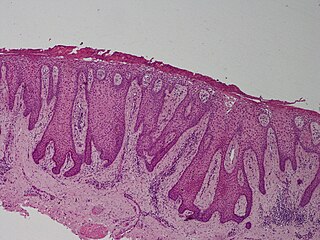
Clear cell acanthoma is a benign clinical and histological lesion initially described as neoplastic, which some authors now regard as a reactive dermatosis. It usually presents as a moist solitary firm, brown-red, well-circumscribed, 5 mm to 2 cm nodule or plaque on the lower extremities of middle-aged to elderly individuals. The lesion has a crusted, scaly peripheral collarette and vascular puncta on the surface. It is characterized by slow growth, and may persist for years. The clinical differential diagnosis includes: dermatofibroma, inflamed seborrheic keratosis, pyogenic granuloma, basal-cell carcinoma, squamous cell carcinoma, verruca vulgaris, psoriatic plaque, and melanoma.

Spiradenomas (SA) are rare, benign cutaneous adnexal tumors that may progress to become their malignant counterparts, i.e. spiradenocarcinomas (SAC). Cutaneous adnexal tumors are a group of skin tumors consisting of tissues that have differentiated towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands. SA and SAC tumors were regarded as eccrine gland tumors and termed eccrine spiradenomas and eccrine spiradenocarcinomas, respectively. However, more recent studies have found them to be hair follicle tumors and commonly term them spiradenomas and spiradenocarcinomas, respectively. Further confusing the situation, SA-like and SAC-like tumors are also 1) manifestations of the inherited disorder, CYLD cutaneous syndrome (CCS), and 2) have repeatedly been confused with an entirely different tumor, adenoid cystic carcinomas of the salivary gland. Here, SA and SAC are strictly defined as sporadic hair follicle tumors that do not include the hereditary CCS spiradenomas and heridtary spiradenocarcinoms of CCS or the adenoid cystic carcinomas.
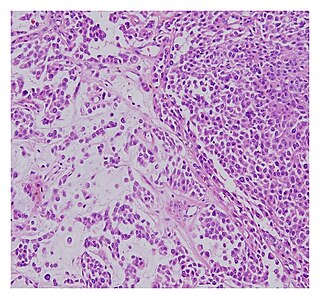
A malignant chondroid syringoma is a very uncommon cutaneous (skin) condition characterised by an adnexal eccrine tumour.
Histiocytic diseases in dogs are a group of diseases in dogs which may involve the skin, and which can be difficult to differentiate from granulomatous, reactive inflammatory or lymphoproliferative diseases. The clinical presentation and behaviour as well as response to therapy vary greatly among the syndromes.

Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) or Reed's syndrome is rare autosomal dominant disorder associated with benign smooth muscle tumors and an increased risk of renal cell carcinoma. It is characterised by multiple cutaneous leiomyomas and, in women, uterine leiomyomas. It predisposes individuals to renal cell cancer, an association denominated hereditary leiomyomatosis and renal cell cancer. It is also associated with increased risk of uterine leiomyosarcoma. The syndrome is caused by a mutation in the fumarate hydratase gene, which leads to an accumulation of fumarate. The inheritance pattern is autosomal dominant and screening can typically begin in childhood.





