
Crystallography is the experimental science of determining the arrangement of atoms in crystalline solids. The word "crystallography" is derived from the Greek words crystallon "cold drop, frozen drop", with its meaning extending to all solids with some degree of transparency, and graphein "to write". In July 2012, the United Nations recognised the importance of the science of crystallography by proclaiming that 2014 would be the International Year of Crystallography.

Structural biology is a branch of molecular biology, biochemistry, and biophysics concerned with the molecular structure of biological macromolecules, how they acquire the structures they have, and how alterations in their structures affect their function. This subject is of great interest to biologists because macromolecules carry out most of the functions of cells, and it is only by coiling into specific three-dimensional shapes that they are able to perform these functions. This architecture, the "tertiary structure" of molecules, depends in a complicated way on each molecule's basic composition, or "primary structure."
The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography, NMR spectroscopy, or, increasingly, cryo-electron microscopy, and submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organisations. The PDB is overseen by an organization called the Worldwide Protein Data Bank, wwPDB.
The Collaborative Computational Project Number 4 in protein crystallography (CCP4) was set up in 1979 in the United Kingdom to support collaboration between researchers working in software development and assemble a comprehensive collection of software for structural biology. The CCP4 core team is located at the Research Complex at Harwell (RCaH) at Rutherford Appleton Laboratory (RAL) in Didcot, near Oxford, UK.
Crystallographic Information File (CIF) is a standard text file format for representing crystallographic information, promulgated by the International Union of Crystallography (IUCr). CIF was developed by the IUCr Working Party on Crystallographic Information in an effort sponsored by the IUCr Commission on Crystallographic Data and the IUCr Commission on Journals. The file format was initially published by Hall, Allen, and Brown and has since been revised, most recently versions 1.1 and 2.0. Full specifications for the format are available at the IUCr website. Many computer programs for molecular viewing are compatible with this format, including Jmol.

Richard Henderson is a Scottish molecular biologist and biophysicist and pioneer in the field of electron microscopy of biological molecules. Henderson shared the Nobel Prize in Chemistry in 2017 with Jacques Dubochet and Joachim Frank.

Jane Shelby Richardson is an American biophysicist best known for developing the Richardson diagram, or ribbon diagram, a method of representing the 3D structure of proteins. Ribbon diagrams have become a standard representation of protein structures that has facilitated further investigation of protein structure and function globally. With interests in astronomy, math, physics, botany, and philosophy, Richardson took an unconventional route to establishing a science career. Today Richardson is a professor in biochemistry at Duke University.
Acta Crystallographica is a series of peer-reviewed scientific journals, with articles centred on crystallography, published by the International Union of Crystallography (IUCr). Originally established in 1948 as a single journal called Acta Crystallographica, there are now six independent Acta Crystallographica titles:

Helen Miriam Berman is a Board of Governors Professor of Chemistry and Chemical Biology at Rutgers University and a former director of the RCSB Protein Data Bank. A structural biologist, her work includes structural analysis of protein-nucleic acid complexes, and the role of water in molecular interactions. She is also the founder and director of the Nucleic Acid Database, and led the Protein Structure Initiative Structural Genomics Knowledgebase.
In biology, a protein structure database is a database that is modeled around the various experimentally determined protein structures. The aim of most protein structure databases is to organize and annotate the protein structures, providing the biological community access to the experimental data in a useful way. Data included in protein structure databases often includes three-dimensional coordinates as well as experimental information, such as unit cell dimensions and angles for x-ray crystallography determined structures. Though most instances, in this case either proteins or a specific structure determinations of a protein, also contain sequence information and some databases even provide means for performing sequence based queries, the primary attribute of a structure database is structural information, whereas sequence databases focus on sequence information, and contain no structural information for the majority of entries. Protein structure databases are critical for many efforts in computational biology such as structure based drug design, both in developing the computational methods used and in providing a large experimental dataset used by some methods to provide insights about the function of a protein.

Foldit is an online puzzle video game about protein folding. It is part of an experimental research project developed by the University of Washington, Center for Game Science, in collaboration with the UW Department of Biochemistry. The objective of Foldit is to fold the structures of selected proteins as perfectly as possible, using tools provided in the game. The highest scoring solutions are analyzed by researchers, who determine whether or not there is a native structural configuration that can be applied to relevant proteins in the real world. Scientists can then use these solutions to target and eradicate diseases and create biological innovations. A 2010 paper in the science journal Nature credited Foldit's 57,000 players with providing useful results that matched or outperformed algorithmically computed solutions.
A crystallographic database is a database specifically designed to store information about the structure of molecules and crystals. Crystals are solids having, in all three dimensions of space, a regularly repeating arrangement of atoms, ions, or molecules. They are characterized by symmetry, morphology, and directionally dependent physical properties. A crystal structure describes the arrangement of atoms, ions, or molecules in a crystal.
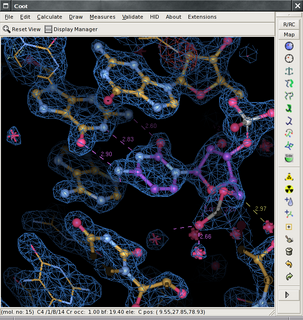
The program Coot is used to display and manipulate atomic models of macromolecules, typically of proteins or nucleic acids, using 3D computer graphics. It is primarily focused on building and validation of atomic models into three-dimensional electron density maps obtained by X-ray crystallography methods, although it has also been applied to data from electron microscopy.
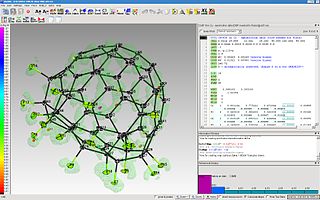
The program ShelXle is a graphical user interface for the structure refinement program SHELXL. ShelXle combines an editor with syntax highlighting for the SHELXL-associated .ins (input) and .res (output) files with an interactive graphical display for visualization of a three-dimensional structure including the electron density (Fo) and difference density (Fo-Fc) maps.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.

Randy John Read is a Wellcome Trust Principal Research Fellow and professor of protein crystallography at the University of Cambridge.

Sjors Hendrik Willem ScheresFRS is a Dutch scientist at the MRC Laboratory of Molecular Biology Cambridge, UK.
Microcrystal electron diffraction, or MicroED, is a CryoEM method that was developed by the Gonen laboratory in late 2013 at the Janelia Research Campus of the Howard Hughes Medical Institute. MicroED is a form of electron crystallography where thin 3D crystals are used for structure determination by electron diffraction.

Mercury is a freeware developed by the Cambridge Crystallographic Data Centre, originally designed as a crystal structure visualization tool. Mercury helps three dimensional visualization of crystal structure and assists in drawing and analysis of crystal packing and intermolecular interactions. Current version Mercury can read "cif", ".mol", ".mol2", ".pdb", ".res", ".sd" and ".xyz" types of files. Mercury has its own file format with filename extension ".mryx".

Władysław Minor also known as Wladek Minor is a Polish-American biophysicist, a specialist in structural biology and protein crystallography. He is a Harrison Distinguished Professor of Molecular Physiology and Biological Physics at the University of Virginia. Minor is a co-author of HKL2000/HKL3000 – crystallographic data processing and structure solution software used to process data and solve structures of macromolecules, as well as small molecules. He is a co-founder of HKL Research, a company that distributes the software. He is also a co-author of a public repository of diffraction images (proteindiffraction.org) for some of the protein structures available in the Protein Data Bank and other software tools for structural biology.
