The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography, NMR spectroscopy, or, increasingly, cryo-electron microscopy, and submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organisations. The PDB is overseen by an organization called the Worldwide Protein Data Bank, wwPDB.
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
A kinemage is an interactive graphic scientific illustration. It often is used to visualize molecules, especially proteins although it can also represent other types of 3-dimensional data. The kinemage system is designed to optimize ease of use, interactive performance, and the perception and communication of detailed 3D information. The kinemage information is stored in a text file, human- and machine-readable, that describes the hierarchy of display objects and their properties, and includes optional explanatory text. The kinemage format is a defined chemical MIME type of 'chemical/x-kinemage' with the file extension '.kin'.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.

Molecular geometry is the three-dimensional arrangement of the atoms that constitute a molecule. It includes the general shape of the molecule as well as bond lengths, bond angles, torsional angles and any other geometrical parameters that determine the position of each atom.

In chemistry, conformational isomerism is a form of stereoisomerism in which the isomers can be interconverted just by rotations about formally single bonds. While any two arrangements of atoms in a molecule that differ by rotation about single bonds can be referred to as different conformations, conformations that correspond to local minima on the potential energy surface are specifically called conformational isomers or conformers. Conformations that correspond to local maxima on the energy surface are the transition states between the local-minimum conformational isomers. Rotations about single bonds involve overcoming a rotational energy barrier to interconvert one conformer to another. If the energy barrier is low, there is free rotation and a sample of the compound exists as a rapidly equilibrating mixture of multiple conformers; if the energy barrier is high enough then there is restricted rotation, a molecule may exist for a relatively long time period as a stable rotational isomer or rotamer. When the time scale for interconversion is long enough for isolation of individual rotamers, the isomers are termed atropisomers. The ring-flip of substituted cyclohexanes constitutes another common form of conformational isomerism.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.

In the context of chemistry and molecular modelling, a force field is a computational method that is used to estimate the forces between atoms within molecules and also between molecules. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or coarse-grained particles in molecular mechanics, molecular dynamics, or Monte Carlo simulations. The parameters for a chosen energy function may be derived from experiments in physics and chemistry, calculations in quantum mechanics, or both. Force fields are interatomic potentials and utilize the same concept as force fields in classical physics, with the difference that the force field parameters in chemistry describe the energy landscape, from which the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.
Internal Coordinate Mechanics (ICM) is a software program and algorithm to predict low-energy conformations of molecules by sampling the space of internal coordinates defining molecular geometry. In ICM each molecule is constructed as a tree from an entry atom where each next atom is built iteratively from the preceding three atoms via three internal variables. The rings kept rigid or imposed via additional restraints. ICM is used for modelling peptides and interactions with substrates and coenzymes.
The Protein Data Bank (pdb) file format is a textual file format describing the three-dimensional structures of molecules held in the Protein Data Bank. The pdb format accordingly provides for description and annotation of protein and nucleic acid structures including atomic coordinates, secondary structure assignments, as well as atomic connectivity. In addition experimental metadata are stored. PDB format is the legacy file format for the Protein Data Bank which now keeps data on biological macromolecules in the newer mmCIF file format.
Molecular graphics is the discipline and philosophy of studying molecules and their properties through graphical representation. IUPAC limits the definition to representations on a "graphical display device". Ever since Dalton's atoms and Kekulé's benzene, there has been a rich history of hand-drawn atoms and molecules, and these representations have had an important influence on modern molecular graphics.
A molecular model is a physical model of an atomistic system that represents molecules and their processes. They play an important role in understanding chemistry and generating and testing hypotheses. The creation of mathematical models of molecular properties and behavior is referred to as molecular modeling, and their graphical depiction is referred to as molecular graphics.
The Collaborative Computational Project Number 4 in protein crystallography (CCP4) was set up in 1979 in the United Kingdom to support collaboration between researchers working in software development and assemble a comprehensive collection of software for structural biology. The CCP4 core team is located at the Research Complex at Harwell (RCaH) at Rutherford Appleton Laboratory (RAL) in Didcot, near Oxford, UK.
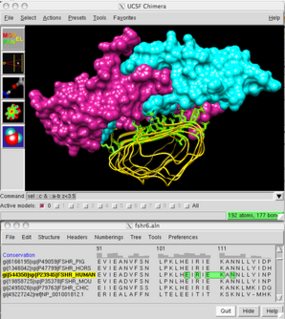
UCSF Chimera is an extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. High-quality images and movies can be created. Chimera includes complete documentation and can be downloaded free of charge for noncommercial use.

Frederic Middlebrook Richards, commonly referred to as Fred Richards, was an American biochemist and biophysicist known for solving the pioneering crystal structure of the ribonuclease S enzyme in 1967 and for defining the concept of solvent-accessible surface. He contributed many key experimental and theoretical results and developed new methods, garnering over 20,000 journal citations in several quite distinct research areas. In addition to the protein crystallography and biochemistry of ribonuclease S, these included solvent accessibility and internal packing of proteins, the first side-chain rotamer library, high-pressure crystallography, new types of chemical tags such as biotin/avidin, the nuclear magnetic resonance (NMR) chemical shift index, and structural and biophysical characterization of the effects of mutations.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.

Paul Emsley is a British crystallographer at the MRC Laboratory of Molecular Biology in Cambridge. He works as an independent scientist and is a member of the Computational Crystallography Group headed by Garib Murshudov.
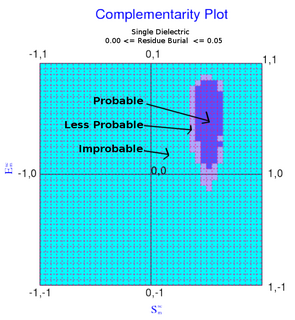
The complementarity plot (CP) is a graphical tool for structural validation of atomic models for both folded globular proteins and protein-protein interfaces. It is based on a probabilistic representation of preferred amino acid side-chain orientation, analogous to the preferred backbone orientation of Ramachandran plots). It can potentially serve to elucidate protein folding as well as binding. The upgraded versions of the software suite is available and maintained in github for both folded globular proteins as well as inter-protein complexes. The software is included in the bioinformatic tool suites OmicTools and Delphi tools.

In biochemistry, a backbone-dependent rotamer library provides the frequencies, mean dihedral angles, and standard deviations of the discrete conformations of the amino acid side chains in proteins as a function of the backbone dihedral angles φ and ψ of the Ramachandran map. By contrast, backbone-independent rotamer libraries express the frequencies and mean dihedral angles for all side chains in proteins, regardless of the backbone conformation of each residue type. Backbone-dependent rotamer libraries have been shown to have significant advantages over backbone-independent rotamer libraries, principally when used as an energy term, by speeding up search times of side-chain packing algorithms used in protein structure prediction and protein design.





