In biology, phylogenetics is the study of the evolutionary history and relationships among or within groups of organisms. These relationships are determined by phylogenetic inference, methods that focus on observed heritable traits, such as DNA sequences, protein amino acid sequences, or morphology. The result of such an analysis is a phylogenetic tree—a diagram containing a hypothesis of relationships that reflects the evolutionary history of a group of organisms.
A phylogenetic tree, phylogeny or evolutionary tree is a graphical representation which shows the evolutionary history between a set of species or taxa during a specific time. In other words, it is a branching diagram or a tree showing the evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical or genetic characteristics. In evolutionary biology, all life on Earth is theoretically part of a single phylogenetic tree, indicating common ancestry. Phylogenetics is the study of phylogenetic trees. The main challenge is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of species or taxa. Computational phylogenetics focuses on the algorithms involved in finding optimal phylogenetic tree in the phylogenetic landscape.
The molecular clock is a figurative term for a technique that uses the mutation rate of biomolecules to deduce the time in prehistory when two or more life forms diverged. The biomolecular data used for such calculations are usually nucleotide sequences for DNA, RNA, or amino acid sequences for proteins.
In phylogenetics and computational phylogenetics, maximum parsimony is an optimality criterion under which the phylogenetic tree that minimizes the total number of character-state changes. Under the maximum-parsimony criterion, the optimal tree will minimize the amount of homoplasy. In other words, under this criterion, the shortest possible tree that explains the data is considered best. Some of the basic ideas behind maximum parsimony were presented by James S. Farris in 1970 and Walter M. Fitch in 1971.
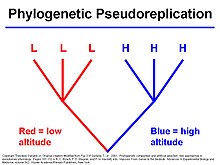
In phylogenetics, long branch attraction (LBA) is a form of systematic error whereby distantly related lineages are incorrectly inferred to be closely related. LBA arises when the amount of molecular or morphological change accumulated within a lineage is sufficient to cause that lineage to appear similar to another long-branched lineage, solely because they have both undergone a large amount of change, rather than because they are related by descent. Such bias is more common when the overall divergence of some taxa results in long branches within a phylogeny. Long branches are often attracted to the base of a phylogenetic tree, because the lineage included to represent an outgroup is often also long-branched. The frequency of true LBA is unclear and often debated, and some authors view it as untestable and therefore irrelevant to empirical phylogenetic inference. Although often viewed as a failing of parsimony-based methodology, LBA could in principle result from a variety of scenarios and be inferred under multiple analytical paradigms.

Joseph "Joe" Felsenstein is a Professor Emeritus in the Departments of Genome Sciences and Biology at the University of Washington in Seattle. He is best known for his work on phylogenetic inference, and is the author of Inferring Phylogenies, and principal author and distributor of the package of phylogenetic inference programs called PHYLIP. Closely related to his work on phylogenetic inference is his introduction of methods for making statistically independent comparisons using phylogenies.
Computational phylogenetics, phylogeny inference, or phylogenetic inference focuses on computational and optimization algorithms, heuristics, and approaches involved in phylogenetic analyses. The goal is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of genes, species, or taxa. Maximum likelihood, parsimony, Bayesian, and minimum evolution are typical optimality criteria used to assess how well a phylogenetic tree topology describes the sequence data. Nearest Neighbour Interchange (NNI), Subtree Prune and Regraft (SPR), and Tree Bisection and Reconnection (TBR), known as tree rearrangements, are deterministic algorithms to search for optimal or the best phylogenetic tree. The space and the landscape of searching for the optimal phylogenetic tree is known as phylogeny search space.
Ancestral reconstruction is the extrapolation back in time from measured characteristics of individuals, populations, or specie to their common ancestors. It is an important application of phylogenetics, the reconstruction and study of the evolutionary relationships among individuals, populations or species to their ancestors. In the context of evolutionary biology, ancestral reconstruction can be used to recover different kinds of ancestral character states of organisms that lived millions of years ago. These states include the genetic sequence, the amino acid sequence of a protein, the composition of a genome, a measurable characteristic of an organism (phenotype), and the geographic range of an ancestral population or species. This is desirable because it allows us to examine parts of phylogenetic trees corresponding to the distant past, clarifying the evolutionary history of the species in the tree. Since modern genetic sequences are essentially a variation of ancient ones, access to ancient sequences may identify other variations and organisms which could have arisen from those sequences. In addition to genetic sequences, one might attempt to track the changing of one character trait to another, such as fins turning to legs.
Biological constraints are factors which make populations resistant to evolutionary change. One proposed definition of constraint is "A property of a trait that, although possibly adaptive in the environment in which it originally evolved, acts to place limits on the production of new phenotypic variants." Constraint has played an important role in the development of such ideas as homology and body plans.
Bayesian inference of phylogeny combines the information in the prior and in the data likelihood to create the so-called posterior probability of trees, which is the probability that the tree is correct given the data, the prior and the likelihood model. Bayesian inference was introduced into molecular phylogenetics in the 1990s by three independent groups: Bruce Rannala and Ziheng Yang in Berkeley, Bob Mau in Madison, and Shuying Li in University of Iowa, the last two being PhD students at the time. The approach has become very popular since the release of the MrBayes software in 2001, and is now one of the most popular methods in molecular phylogenetics.

Evolutionary physiology is the study of the biological evolution of physiological structures and processes; that is, the manner in which the functional characteristics of individuals in a population of organisms have responded to natural selection across multiple generations during the history of the population. It is a sub-discipline of both physiology and evolutionary biology. Practitioners in the field come from a variety of backgrounds, including physiology, evolutionary biology, ecology, and genetics.
Paul H. Harvey is a British evolutionary biologist. He is Professor of Zoology and was head of the zoology department at the University of Oxford from 1998 to 2011 and Secretary of the Zoological Society of London from 2000 to 2011, holding these posts in conjunction with a professorial fellowship at Jesus College, Oxford.
Theodore Garland Jr. is a biologist specializing in evolutionary physiology at the University of California, Riverside.
Ziheng Yang FRS is a Chinese biologist. He holds the R.A. Fisher Chair of Statistical Genetics at University College London, and is the Director of R.A. Fisher Centre for Computational Biology at UCL. He was elected a Fellow of the Royal Society in 2006.
Mark David Pagel FRS is an evolutionary biologist and professor. He heads the Evolutionary Biology Group at the University of Reading. He is known for comparative studies in evolutionary biology. In 1994, with his spouse, anthropologist Ruth Mace, Pagel pioneered the Comparative Method in Anthropology.
Microbial phylogenetics is the study of the manner in which various groups of microorganisms are genetically related. This helps to trace their evolution. To study these relationships biologists rely on comparative genomics, as physiology and comparative anatomy are not possible methods.
The term phylogenetic niche conservatism has seen increasing use in recent years in the scientific literature, though the exact definition has been a matter of some contention. Fundamentally, phylogenetic niche conservatism refers to the tendency of species to retain their ancestral traits. When defined as such, phylogenetic niche conservatism is therefore nearly synonymous with phylogenetic signal. The point of contention is whether or not "conservatism" refers simply to the tendency of species to resemble their ancestors, or implies that "closely related species are more similar than expected based on phylogenetic relationships". If the latter interpretation is employed, then phylogenetic niche conservatism can be seen as an extreme case of phylogenetic signal, and implies that the processes which prevent divergence are in operation in the lineage under consideration. Despite efforts by Jonathan Losos to end this habit, however, the former interpretation appears to frequently motivate scientific research. In this case, phylogenetic niche conservatism might best be considered a form of phylogenetic signal reserved for traits with broad-scale ecological ramifications. Thus, phylogenetic niche conservatism is usually invoked with regards to closely related species occurring in similar environments.
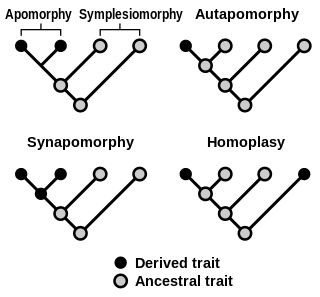
Homoplasy, in biology and phylogenetics, is the term used to describe a feature that has been gained or lost independently in separate lineages over the course of evolution. This is different from homology, which is the term used to characterize the similarity of features that can be parsimoniously explained by common ancestry. Homoplasy can arise from both similar selection pressures acting on adapting species, and the effects of genetic drift.

Phylogenetic signal is an evolutionary and ecological term, that describes the tendency or the pattern of related biological species to resemble each other more than any other species that is randomly picked from the same phylogenetic tree.