
Blood alcohol content (BAC), also called blood alcohol concentration or blood alcohol level, is a measurement of alcohol intoxication used for legal or medical purposes.

In vitro studies are performed with microorganisms, cells, or biological molecules outside their normal biological context. Colloquially called "test-tube experiments", these studies in biology and its subdisciplines are traditionally done in labware such as test tubes, flasks, Petri dishes, and microtiter plates. Studies conducted using components of an organism that have been isolated from their usual biological surroundings permit a more detailed or more convenient analysis than can be done with whole organisms; however, results obtained from in vitro experiments may not fully or accurately predict the effects on a whole organism. In contrast to in vitro experiments, in vivo studies are those conducted in living organisms, including humans, known as clinical trials, and whole plants.
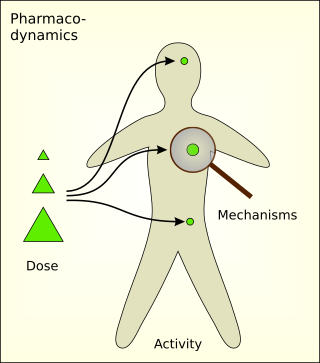
Pharmacodynamics (PD) is the study of the biochemical and physiologic effects of drugs. The effects can include those manifested within animals, microorganisms, or combinations of organisms.
In pharmacology, bioavailability is a subcategory of absorption and is the fraction (%) of an administered drug that reaches the systemic circulation.
In the physical sciences, a partition coefficient (P) or distribution coefficient (D) is the ratio of concentrations of a compound in a mixture of two immiscible solvents at equilibrium. This ratio is therefore a comparison of the solubilities of the solute in these two liquids. The partition coefficient generally refers to the concentration ratio of un-ionized species of compound, whereas the distribution coefficient refers to the concentration ratio of all species of the compound.
Compartmental models are a very general modelling technique. They are often applied to the mathematical modelling of infectious diseases. The population is assigned to compartments with labels – for example, S, I, or R,. People may progress between compartments. The order of the labels usually shows the flow patterns between the compartments; for example SEIS means susceptible, exposed, infectious, then susceptible again.
In pharmacology, the volume of distribution is the theoretical volume that would be necessary to contain the total amount of an administered drug at the same concentration that it is observed in the blood plasma. In other words, it is the ratio of amount of drug in a body (dose) to concentration of the drug that is measured in blood, plasma, and un-bound in interstitial fluid.
In pharmacology, clearance is a pharmacokinetic parameter representing the efficiency of drug elimination. This is the rate of elimination of a substance divided by its concentration. The parameter also indicates the theoretical volume of plasma from which a substance would be completely removed per unit time. Usually, clearance is measured in L/h or mL/min. The quantity reflects the rate of drug elimination divided by plasma concentration. Excretion, on the other hand, is a measurement of the amount of a substance removed from the body per unit time. While clearance and excretion of a substance are related, they are not the same thing. The concept of clearance was described by Thomas Addis, a graduate of the University of Edinburgh Medical School.
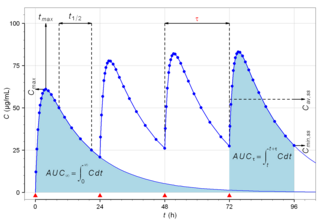
Biological half-life is the time taken for concentration of a biological substance to decrease from its maximum concentration (Cmax) to half of Cmax in the blood plasma. It is denoted by the abbreviation .
Absorption is the journey of a drug travelling from the site of administration to the site of action.
Pharmacokinetics, sometimes abbreviated as PK, is a branch of pharmacology dedicated to describing how the body affects a specific substance after administration. The substances of interest include any chemical xenobiotic such as pharmaceutical drugs, pesticides, food additives, cosmetics, etc. It attempts to analyze chemical metabolism and to discover the fate of a chemical from the moment that it is administered up to the point at which it is completely eliminated from the body. Pharmacokinetics is based on mathematical modeling that places great emphasis on the relationship between drug plasma concentration and the time elapsed since the drug's administration. Pharmacokinetics is the study of how an organism affects the drug, whereas pharmacodynamics (PD) is the study of how the drug affects the organism. Both together influence dosing, benefit, and adverse effects, as seen in PK/PD models.
GNU MCSim is a suite of simulation software. It allows one to design one's own statistical or simulation models, perform Monte Carlo simulations, and Bayesian inference through (tempered) Markov chain Monte Carlo simulations. The latest version allows parallel computing of Monte Carlo or MCMC simulations.
Cmin is a term used in pharmacokinetics for the minimum blood plasma concentration reached by a drug during a dosing interval, which is the time interval between administration of two doses. This definition is slightly different from Ctrough, the concentration immediately prior to administration of the next dose. Cmin is the opposite of Cmax, the maximum concentration that the drug reaches. Cmin must be above certain thresholds, such as the minimum inhibitory concentration (MIC), to achieve a therapeutic effect.
The plateau principle is a mathematical model or scientific law originally developed to explain the time course of drug action (pharmacokinetics). The principle has wide applicability in pharmacology, physiology, nutrition, biochemistry, and system dynamics. It applies whenever a drug or nutrient is infused or ingested at a relatively constant rate and when a constant fraction is eliminated during each time interval. Under these conditions, any change in the rate of infusion leads to an exponential increase or decrease until a new level is achieved. This behavior is also called an approach to steady state because rather than causing an indefinite increase or decrease, a natural balance is achieved when the rate of infusion or production is balanced by the rate of loss.
An organ-on-a-chip (OOC) is a multi-channel 3-D microfluidic cell culture, integrated circuit (chip) that simulates the activities, mechanics and physiological response of an entire organ or an organ system. It constitutes the subject matter of significant biomedical engineering research, more precisely in bio-MEMS. The convergence of labs-on-chips (LOCs) and cell biology has permitted the study of human physiology in an organ-specific context. By acting as a more sophisticated in vitro approximation of complex tissues than standard cell culture, they provide the potential as an alternative to animal models for drug development and toxin testing.
A Logan plot is a graphical analysis technique based on the compartment model that uses linear regression to analyze pharmacokinetics of tracers involving reversible uptake. It is mainly used for the evaluation of nuclear medicine imaging data after the injection of a labeled ligand that binds reversibly to specific receptor or enzyme.
In vitro to in vivo extrapolation (IVIVE) refers to the qualitative or quantitative transposition of experimental results or observations made in vitro to predict phenomena in vivo, biological organisms.
Leon Aarons is an Australian chemist who researches and teaches in the areas of pharmacodynamics and pharmacokinetics. He lives in the United Kingdom and from 1976 has been a professor of pharmacometrics at the University of Manchester. In the interest of promoting the effective development of drugs, the main focus of his work is optimizing pharmacological models, the design of clinical studies, and data analysis and interpretation in the field of population pharmacokinetics. From 1985 to 2010 Aarons was an editor emeritus of the Journal of Pharmacokinetics and Pharmacodynamics and is a former executive editor of the British Journal of Clinical Pharmacology.
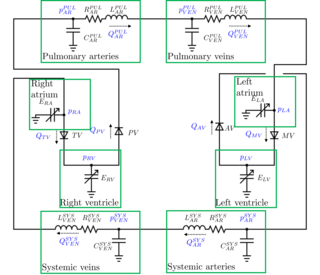
A lumped parameter cardiovascular model is a zero-dimensional mathematical model used to describe the hemodynamics of the cardiovascular system. Given a set of parameters that have a physical meaning, it allows to study the changes in blood pressures or flow rates throughout the cardiovascular system. Modifying the parameters, it is possible to study the effects of a specific disease. For example, arterial hypertension is modeled increasing the arterial resistances of the model.
In medicinal chemistry, Drug Permeability is an empirical parameter that indicates how quickly a chemical entity or an active pharmaceutical ingredient crosses a biological membrane or another biological barrier to become bioavailable in the body. Drug permeability, together with drug aqueous solubility are the two parameters which defines the fate of the active ingredient after oral administration that ultimately defines its bioavailability. When drug permeability is empirically measured in vitro, it is generally called apparent permeability (Papp) as its absolute value varies according to the method selected for its measurement. Papp is measured in vitro utilizing cellular based barriers such as the Caco-2 model or utilizing artificial biomimetic barriers, such as the Parallel Artificial Membrane Permeation Assay (PAMPA) or the PermeaPad. All these methods are built on an acceptor compartment where the drug solution is placed, a biomimetic barrier and an acceptor compartment, where the drug concentration is quantified over time. By maintaining sink condition, a steady state is reached after a lag time.
![Graphic representation of a physiologically based whole body model. Here, it is dissected into seven tissue/organ compartments: brain, lungs and heart, pancreas, liver, gut, kidney and adipose/muscle tissue. Blood flows, Q, and concentration, [X], of a substance of interest are depicted. WholeBody wiki.svg](http://upload.wikimedia.org/wikipedia/commons/thumb/2/22/WholeBody_wiki.svg/220px-WholeBody_wiki.svg.png)




















