Proteasomes are protein complexes which degrade unneeded or damaged proteins by proteolysis, a chemical reaction that breaks peptide bonds. Enzymes that help such reactions are called proteases.

Protein folding is the physical process by which a protein, after synthesis by a ribosome as a linear chain of amino acids, changes from an unstable random coil into a more ordered three-dimensional structure. This structure permits the protein to become biologically functional.

In molecular biology, molecular chaperones are proteins that assist the conformational folding or unfolding of large proteins or macromolecular protein complexes. There are a number of classes of molecular chaperones, all of which function to assist large proteins in proper protein folding during or after synthesis, and after partial denaturation. Chaperones are also involved in the translocation of proteins for proteolysis.
Heat shock proteins (HSPs) are a family of proteins produced by cells in response to exposure to stressful conditions. They were first described in relation to heat shock, but are now known to also be expressed during other stresses including exposure to cold, UV light and during wound healing or tissue remodeling. Many members of this group perform chaperone functions by stabilizing new proteins to ensure correct folding or by helping to refold proteins that were damaged by the cell stress. This increase in expression is transcriptionally regulated. The dramatic upregulation of the heat shock proteins is a key part of the heat shock response and is induced primarily by heat shock factor (HSF). HSPs are found in virtually all living organisms, from bacteria to humans.
The 70 kilodalton heat shock proteins are a family of conserved ubiquitously expressed heat shock proteins. Proteins with similar structure exist in virtually all living organisms. Intracellularly localized Hsp70s are an important part of the cell's machinery for protein folding, performing chaperoning functions, and helping to protect cells from the adverse effects of physiological stresses. Additionally, membrane-bound Hsp70s have been identified as a potential target for cancer therapies and their extracellularly localized counterparts have been identified as having both membrane-bound and membrane-free structures.
HSP60, also known as chaperonins (Cpn), is a family of heat shock proteins originally sorted by their 60kDa molecular mass. They prevent misfolding of proteins during stressful situations such as high heat, by assisting protein folding. HSP60 belong to a large class of molecules that assist protein folding, called molecular chaperones.
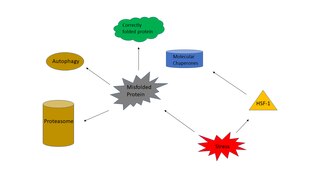
The heat shock response (HSR) is a cell stress response that increases the number of molecular chaperones to combat the negative effects on proteins caused by stressors such as increased temperatures, oxidative stress, and heavy metals. In a normal cell, proteostasis must be maintained because proteins are the main functional units of the cell. Many proteins take on a defined configuration in a process known as protein folding in order to perform their biological functions. If these structures are altered, critical processes could be affected, leading to cell damage or death. The heat shock response can be employed under stress to induce the expression of heat shock proteins (HSP), many of which are molecular chaperones, that help prevent or reverse protein misfolding and provide an environment for proper folding.

Endoplasmic-reticulum-associated protein degradation (ERAD) designates a cellular pathway which targets misfolded proteins of the endoplasmic reticulum for ubiquitination and subsequent degradation by a protein-degrading complex, called the proteasome.
In eukaryotic cells, an aggresome refers to an aggregation of misfolded proteins in the cell, formed when the protein degradation system of the cell is overwhelmed. Aggresome formation is a highly regulated process that possibly serves to organize misfolded proteins into a single location.
The unfolded protein response (UPR) is a cellular stress response related to the endoplasmic reticulum (ER) stress. It has been found to be conserved between mammalian species, as well as yeast and worm organisms.

Heat shock 70 kDa protein 1, also termed Hsp72, is a protein that in humans is encoded by the HSPA1A gene. As a member of the heat shock protein 70 family and a chaperone protein, it facilitates the proper folding of newly translated and misfolded proteins, as well as stabilize or degrade mutant proteins. In addition, Hsp72 also facilitates DNA repair. Its functions contribute to biological processes including signal transduction, apoptosis, protein homeostasis, and cell growth and differentiation. It has been associated with an extensive number of cancers, neurodegenerative diseases, cell senescence and aging, and inflammatory diseases such as Diabetes mellitus type 2 and rheumatoid arthritis.

26S proteasome non-ATPase regulatory subunit 4, also as known as 26S Proteasome Regulatory Subunit Rpn10, is an enzyme that in humans is encoded by the PSMD4 gene. This protein is one of the 19 essential subunits that contributes to the complete assembly of 19S proteasome complex.

26S protease regulatory subunit 4, also known as 26S proteasome AAA-ATPase subunit Rpt2, is an enzyme that in humans is encoded by the PSMC1 gene. This protein is one of the 19 essential subunits of a complete assembled 19S proteasome complex. Six 26S proteasome AAA-ATPase subunits together with four non-ATPase subunits form the base sub complex of 19S regulatory particle for proteasome complex.

26S proteasome non-ATPase regulatory subunit 7, also known as 26S proteasome non-ATPase subunit Rpn8, is an enzyme that in humans is encoded by the PSMD7 gene.

26S proteasome non-ATPase regulatory subunit 1, also as known as 26S Proteasome Regulatory Subunit Rpn2, is a protein that in humans is encoded by the PSMD1 gene. This protein is one of the 19 essential subunits that contributes to the complete assembly of 19S proteasome complex.
26S proteasome non-ATPase regulatory subunit 14, also known as 26S proteasome non-ATPase subunit Rpn11, is an enzyme that in humans is encoded by the PSMD14 gene. This protein is one of the 19 essential subunits of the complete assembled 19S proteasome complex. Nine subunits Rpn3, Rpn5, Rpn6, Rpn7, Rpn8, Rpn9, Rpn11, SEM1, and Rpn12 form the lid sub complex of the 19S regulatory particle of the proteasome complex.
Proteostasis is the dynamic regulation of a balanced, functional proteome. The proteostasis network includes competing and integrated biological pathways within cells that control the biogenesis, folding, trafficking, and degradation of proteins present within and outside the cell. Loss of proteostasis is central to understanding the cause of diseases associated with excessive protein misfolding and degradation leading to loss-of-function phenotypes, as well as aggregation-associated degenerative disorders. Therapeutic restoration of proteostasis may treat or resolve these pathologies.
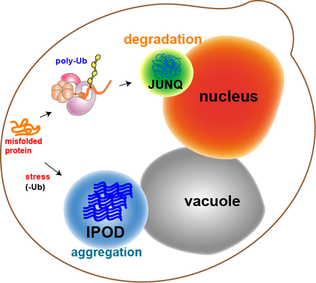
JUNQ and IPOD are types of cytosolic protein inclusion bodies in eukaryotes.
Chemical chaperones are a class of small molecules that function to enhance the folding and/or stability of proteins. Chemical chaperones are a broad and diverse group of molecules, and they can influence protein stability and polypeptide organization through a variety of mechanisms. Chemical chaperones are used for a range of applications, from production of recombinant proteins to treatment of protein misfolding in vivo.
Chaperones, also called molecular chaperones, are proteins that assist other proteins in assuming their three-dimensional fold, which is necessary for protein function. However, the fold of a protein is sensitive to environmental conditions, such as temperature and pH, and thus chaperones are needed to keep proteins in their functional fold across various environmental conditions. Chaperones are an integral part of a cell's protein quality control network by assisting in protein folding and are ubiquitous across diverse biological taxa. Since protein folding, and therefore protein function, is susceptible to environmental conditions, chaperones could represent an important cellular aspect of biodiversity and environmental tolerance by organisms living in hazardous conditions. Chaperones also affect the evolution of proteins in general, as many proteins fundamentally require chaperones to fold or are naturally prone to misfolding, and therefore mitigates protein aggregation.