Crystallography is the experimental science of determining the arrangement of atoms in crystalline solids. The word "crystallography" is derived from the Greek words crystallon "cold drop, frozen drop", with its meaning extending to all solids with some degree of transparency, and graphein "to write". In July 2012, the United Nations recognised the importance of the science of crystallography by proclaiming that 2014 would be the International Year of Crystallography.

X-ray crystallography (XRC) is the experimental science determining the atomic and molecular structure of a crystal, in which the crystalline structure causes a beam of incident X-rays to diffract into many specific directions. By measuring the angles and intensities of these diffracted beams, a crystallographer can produce a three-dimensional picture of the density of electrons within the crystal. From this electron density, the mean positions of the atoms in the crystal can be determined, as well as their chemical bonds, their crystallographic disorder, and various other information.

A chemical structure determination includes a chemist's specifying the molecular geometry and, when feasible and necessary, the electronic structure of the target molecule or other solid. Molecular geometry refers to the spatial arrangement of atoms in a molecule and the chemical bonds that hold the atoms together, and can be represented using structural formulae and by molecular models; complete electronic structure descriptions include specifying the occupation of a molecule's molecular orbitals. Structure determination can be applied to a range of targets from very simple molecules, to very complex ones.
In physics, the phase problem is the problem of loss of information concerning the phase that can occur when making a physical measurement. The name comes from the field of X-ray crystallography, where the phase problem has to be solved for the determination of a structure from diffraction data. The phase problem is also met in the fields of imaging and signal processing. Various approaches of phase retrieval have been developed over the years.
Molecular graphics (MG) is the discipline and philosophy of studying molecules and their properties through graphical representation. IUPAC limits the definition to representations on a "graphical display device". Ever since Dalton's atoms and Kekulé's benzene, there has been a rich history of hand-drawn atoms and molecules, and these representations have had an important influence on modern molecular graphics. This article concentrates on the use of computers to create molecular graphics. Note, however, that many molecular graphics programs and systems have close coupling between the graphics and editing commands or calculations such as in molecular modelling.
Multi-wavelength anomalous diffraction is a technique used in X-ray crystallography that facilitates the determination of the three-dimensional structure of biological macromolecules via solution of the phase problem.
Modeller, often stylized as MODELLER, is a computer program used for homology modeling to produce models of protein tertiary structures and quaternary structures (rarer). It implements a method inspired by nuclear magnetic resonance spectroscopy of proteins, termed satisfaction of spatial restraints, by which a set of geometrical criteria are used to create a probability density function for the location of each atom in the protein. The method relies on an input sequence alignment between the target amino acid sequence to be modeled and a template protein which structure has been solved.
Acta Crystallographica is a series of peer-reviewed scientific journals, with articles centred on crystallography, published by the International Union of Crystallography (IUCr). Originally established in 1948 as a single journal called Acta Crystallographica, there are now six independent Acta Crystallographica titles:
Multiple isomorphous replacement (MIR) is historically the most common approach to solving the phase problem in X-ray crystallography studies of proteins. For protein crystals this method is conducted by soaking the crystal of a sample to be analyzed with a heavy atom solution or co-crystallization with the heavy atom. The addition of the heavy atom to the structure should not affect the crystal formation or unit cell dimensions in comparison to its native form, hence, they should be isomorphic.
Single-wavelength anomalous diffraction (SAD) is a technique used in X-ray crystallography that facilitates the determination of the structure of proteins or other biological macromolecules by allowing the solution of the phase problem. In contrast to multi-wavelength anomalous diffraction, SAD uses a single dataset at a single appropriate wavelength. One advantage of the technique is the minimization of time spent in the beam by the crystal, thus reducing potential radiation damage to the molecule while collecting data. SAD is sometimes called "single-wavelength anomalous dispersion", but no dispersive differences are used in this technique since the data are collected at a single wavelength. Today, selenium-SAD is commonly used for experimental phasing due to the development of methods for selenomethionine incorporation into recombinant proteins.
A crystallographic database is a database specifically designed to store information about the structure of molecules and crystals. Crystals are solids having, in all three dimensions of space, a regularly repeating arrangement of atoms, ions, or molecules. They are characterized by symmetry, morphology, and directionally dependent physical properties. A crystal structure describes the arrangement of atoms, ions, or molecules in a crystal.
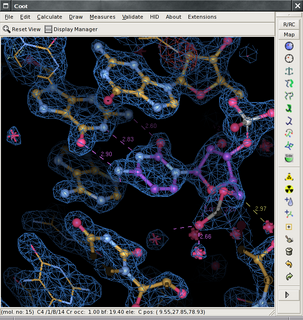
The program Coot is used to display and manipulate atomic models of macromolecules, typically of proteins or nucleic acids, using 3D computer graphics. It is primarily focused on building and validation of atomic models into three-dimensional electron density maps obtained by X-ray crystallography methods, although it has also been applied to data from electron microscopy.
Olex and Olex2 are versatile software for crystallographic research. Olex used to be a research project developed during PhD to implement topological analysis of polymeric chemical structures and still is widely used around the world. Olex2 is an open source project with the C++ code portable to Windows, Mac and Linux. Although the projects share the common name they are not related at the source code level.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.

Randy John Read is a Wellcome Trust Principal Research Fellow and Professor of Protein Crystallography at the University of Cambridge.
George Michael Sheldrick, FRS is a British chemist who specialises in molecular structure determination. He is one of the most cited workers in the field, having over 280,000 citations as of 2020 and an h-index of 113. He was a professor at the University of Göttingen from 1978 until his retirement in 2011.
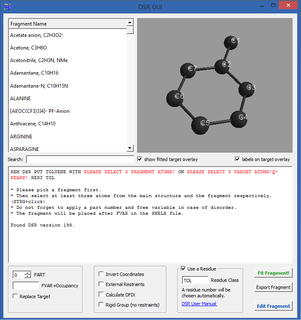
The Disordered Structure Refinement program (DSR), written by Daniel Kratzert, is designed to simplify the modeling of molecular disorder in crystal structures using SHELXL by George M. Sheldrick. It has a database of approximately 120 standard solvent molecules and molecular moieties. These can be inserted into the crystal structure with little effort, while at the same time chemically meaningful binding and angular restraints are set. DSR was developed because the previous description of disorder in crystal structures with SHELXL was very lengthy and error-prone. Instead of editing large text files manually and defining restraints manually, this process is automated with DSR.
The Multipole Density Formalism is an X-ray crystallography method of electron density modelling proposed by Niels K. Hansen and Philip Coppens in 1978. Unlike the commonly used Independent Atom Model, the Hansen-Coppens Formalism presents an aspherical approach, allowing one to model the electron distribution around a nucleus separately in different directions and therefore describe numerous chemical features of a molecule inside the unit cell of an examined crystal in detail.
Quantum crystallography is a branch of crystallography that investigates crystalline materials within the framework of quantum mechanics, with analysis and representation, in position or in momentum space, of quantities like wave function, electron charge and spin density, density matrices and all properties related to them. Like the quantum chemistry, Quantum crystallography involves both experimental and computational work. The theoretical part of quantum crystallography is based on quantum mechanical calculations of atomic/molecular/crystal wave functions, density matrices or density models, used to simulate the electronic structure of a crystalline material. While in quantum chemistry, the experimental works mainly rely on spectroscopy, in quantum crystallography the scattering techniques play the central role, although spectroscopy as well as atomic microscopy are also sources of information.

Mercury is a freeware developed by the Cambridge Crystallographic Data Centre, originally designed as a crystal structure visualization tool. Mercury helps three dimensional visualization of crystal structure and assists in drawing and analysis of crystal packing and intermolecular interactions. Current version Mercury can read "cif", ".mol", ".mol2", ".pdb", ".res", ".sd" and ".xyz" types of files. Mercury has its own file format with filename extension ".mryx".