Bra–ket notation, also called Dirac notation, is a notation for linear algebra and linear operators on complex vector spaces together with their dual space both in the finite-dimensional and infinite-dimensional case. It is specifically designed to ease the types of calculations that frequently come up in quantum mechanics. Its use in quantum mechanics is quite widespread.
In quantum mechanics, the Hamiltonian of a system is an operator corresponding to the total energy of that system, including both kinetic energy and potential energy. Its spectrum, the system's energy spectrum or its set of energy eigenvalues, is the set of possible outcomes obtainable from a measurement of the system's total energy. Due to its close relation to the energy spectrum and time-evolution of a system, it is of fundamental importance in most formulations of quantum theory.
In quantum chemistry and molecular physics, the Born–Oppenheimer (BO) approximation is the best-known mathematical approximation in molecular dynamics. Specifically, it is the assumption that the wave functions of atomic nuclei and electrons in a molecule can be treated separately, based on the fact that the nuclei are much heavier than the electrons. Due to the larger relative mass of a nucleus compared to an electron, the coordinates of the nuclei in a system are approximated as fixed, while the coordinates of the electrons are dynamic. The approach is named after Max Born and his 23-year-old graduate student J. Robert Oppenheimer, the latter of whom proposed it in 1927 during a period of intense ferment in the development of quantum mechanics.
In physics, an operator is a function over a space of physical states onto another space of physical states. The simplest example of the utility of operators is the study of symmetry. Because of this, they are useful tools in classical mechanics. Operators are even more important in quantum mechanics, where they form an intrinsic part of the formulation of the theory.
The adiabatic theorem is a concept in quantum mechanics. Its original form, due to Max Born and Vladimir Fock (1928), was stated as follows:
In physics, a free particle is a particle that, in some sense, is not bound by an external force, or equivalently not in a region where its potential energy varies. In classical physics, this means the particle is present in a "field-free" space. In quantum mechanics, it means the particle is in a region of uniform potential, usually set to zero in the region of interest since the potential can be arbitrarily set to zero at any point in space.

In quantum mechanics, a two-state system is a quantum system that can exist in any quantum superposition of two independent quantum states. The Hilbert space describing such a system is two-dimensional. Therefore, a complete basis spanning the space will consist of two independent states. Any two-state system can also be seen as a qubit.
Vibronic coupling in a molecule involves the interaction between electronic and nuclear vibrational motion. The term "vibronic" originates from the combination of the terms "vibrational" and "electronic", denoting the idea that in a molecule, vibrational and electronic interactions are interrelated and influence each other. The magnitude of vibronic coupling reflects the degree of such interrelation.
One of the guiding principles in modern chemical dynamics and spectroscopy is that the motion of the nuclei in a molecule is slow compared to that of its electrons. This is justified by the large disparity between the mass of an electron, and the typical mass of a nucleus and leads to the Born–Oppenheimer approximation and the idea that the structure and dynamics of a chemical species are largely determined by nuclear motion on potential energy surfaces.
The Ehrenfest theorem, named after Austrian theoretical physicist Paul Ehrenfest, relates the time derivative of the expectation values of the position and momentum operators x and p to the expectation value of the force on a massive particle moving in a scalar potential ,
The Born–Huang approximation is an approximation closely related to the Born–Oppenheimer approximation. It takes into account diagonal nonadiabatic effects in the electronic Hamiltonian better than the Born–Oppenheimer approximation. Despite the addition of correction terms, the electronic states remain uncoupled under the Born–Huang approximation, making it an adiabatic approximation.
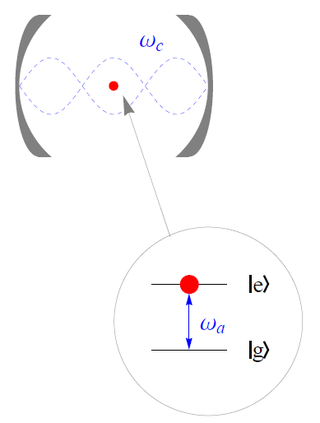
The Jaynes–Cummings model is a theoretical model in quantum optics. It describes the system of a two-level atom interacting with a quantized mode of an optical cavity, with or without the presence of light. It was originally developed to study the interaction of atoms with the quantized electromagnetic field in order to investigate the phenomena of spontaneous emission and absorption of photons in a cavity.
The theoretical and experimental justification for the Schrödinger equation motivates the discovery of the Schrödinger equation, the equation that describes the dynamics of nonrelativistic particles. The motivation uses photons, which are relativistic particles with dynamics described by Maxwell's equations, as an analogue for all types of particles.

In physics, a quantum vortex represents a quantized flux circulation of some physical quantity. In most cases, quantum vortices are a type of topological defect exhibited in superfluids and superconductors. The existence of quantum vortices was first predicted by Lars Onsager in 1949 in connection with superfluid helium. Onsager reasoned that quantisation of vorticity is a direct consequence of the existence of a superfluid order parameter as a spatially continuous wavefunction. Onsager also pointed out that quantum vortices describe the circulation of superfluid and conjectured that their excitations are responsible for superfluid phase transitions. These ideas of Onsager were further developed by Richard Feynman in 1955 and in 1957 were applied to describe the magnetic phase diagram of type-II superconductors by Alexei Alexeyevich Abrikosov. In 1935 Fritz London published a very closely related work on magnetic flux quantization in superconductors. London's fluxoid can also be viewed as a quantum vortex.
In 1927, a year after the publication of the Schrödinger equation, Hartree formulated what are now known as the Hartree equations for atoms, using the concept of self-consistency that Lindsay had introduced in his study of many electron systems in the context of Bohr theory. Hartree assumed that the nucleus together with the electrons formed a spherically symmetric field. The charge distribution of each electron was the solution of the Schrödinger equation for an electron in a potential , derived from the field. Self-consistency required that the final field, computed from the solutions, was self-consistent with the initial field, and he thus called his method the self-consistent field method.
A flavor of the k·p perturbation theory used for calculating the structure of multiple, degenerate electronic bands in bulk and quantum well semiconductors. The method is a generalization of the single band k·p theory.
An electric dipole transition is the dominant effect of an interaction of an electron in an atom with the electromagnetic field.
In physics, Berry connection and Berry curvature are related concepts which can be viewed, respectively, as a local gauge potential and gauge field associated with the Berry phase or geometric phase. The concept was first introduced by S. Pancharatnam as geometric phase and later elaborately explained and popularized by Michael Berry in a paper published in 1984 emphasizing how geometric phases provide a powerful unifying concept in several branches of classical and quantum physics.
In pure and applied mathematics, quantum mechanics and computer graphics, a tensor operator generalizes the notion of operators which are scalars and vectors. A special class of these are spherical tensor operators which apply the notion of the spherical basis and spherical harmonics. The spherical basis closely relates to the description of angular momentum in quantum mechanics and spherical harmonic functions. The coordinate-free generalization of a tensor operator is known as a representation operator.

Newton-X is a general program for molecular dynamics simulations beyond the Born-Oppenheimer approximation. It has been primarily used for simulations of ultrafast processes in photoexcited molecules. It has also been used for simulation of band envelops of absorption and emission spectra.





























