Genomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.

A DNA sequencer is a scientific instrument used to automate the DNA sequencing process. Given a sample of DNA, a DNA sequencer is used to determine the order of the four bases: G (guanine), C (cytosine), A (adenine) and T (thymine). This is then reported as a text string, called a read. Some DNA sequencers can be also considered optical instruments as they analyze light signals originating from fluorochromes attached to nucleotides.

A nanopore is a pore of nanometer size. It may, for example, be created by a pore-forming protein or as a hole in synthetic materials such as silicon or graphene.

Nanopore sequencing is a third generation approach used in the sequencing of biopolymers — specifically, polynucleotides in the form of DNA or RNA.
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.

SOLiD (Sequencing by Oligonucleotide Ligation and Detection) is a next-generation DNA sequencing technology developed by Life Technologies and has been commercially available since 2006. This next generation technology generates 108 - 109 small sequence reads at one time. It uses 2 base encoding to decode the raw data generated by the sequencing platform into sequence data.
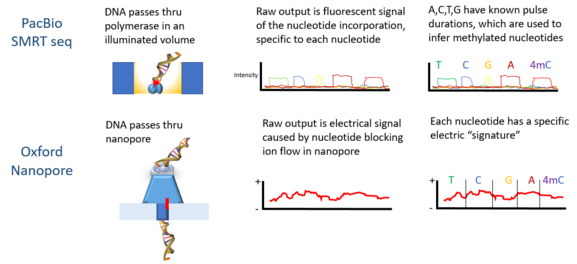
Single-molecule real-time (SMRT) sequencing is a parallelized single molecule DNA sequencing method. Single-molecule real-time sequencing utilizes a zero-mode waveguide (ZMW). A single DNA polymerase enzyme is affixed at the bottom of a ZMW with a single molecule of DNA as a template. The ZMW is a structure that creates an illuminated observation volume that is small enough to observe only a single nucleotide of DNA being incorporated by DNA polymerase. Each of the four DNA bases is attached to one of four different fluorescent dyes. When a nucleotide is incorporated by the DNA polymerase, the fluorescent tag is cleaved off and diffuses out of the observation area of the ZMW where its fluorescence is no longer observable. A detector detects the fluorescent signal of the nucleotide incorporation, and the base call is made according to the corresponding fluorescence of the dye.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.

RNA-Seq is a technique that uses next-generation sequencing to reveal the presence and quantity of RNA molecules in a biological sample, providing a snapshot of gene expression in the sample, also known as transcriptome.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.
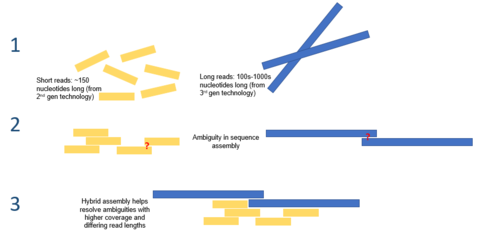
In DNA sequencing, a read is an inferred sequence of base pairs corresponding to all or part of a single DNA fragment. A typical sequencing experiment involves fragmentation of the genome into millions of molecules, which are size-selected and ligated to adapters. The set of fragments is referred to as a sequencing library, which is sequenced to produce a set of reads.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.
Oxford Nanopore Technologies plc is a UK-based company which develops and sells nanopore sequencing products for the direct, electronic analysis of single molecules.
David Wilson Deamer is an American biologist and Research Professor of Biomolecular Engineering at the University of California, Santa Cruz. Deamer has made significant contributions to the field of membrane biophysics. His work led to a novel method of DNA sequencing and a more complete understanding of the role of membranes in the origin of life.

In epitranscriptomic sequencing, most methods focus on either (1) enrichment and purification of the modified RNA molecules before running on the RNA sequencer, or (2) improving or modifying bioinformatics analysis pipelines to call the modification peaks. Most methods have been adapted and optimized for mRNA molecules, except for modified bisulfite sequencing for profiling 5-methylcytidine which was optimized for tRNAs and rRNAs.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.
Clinical metagenomic next-generation sequencing (mNGS) is the comprehensive analysis of microbial and host genetic material in clinical samples from patients by next-generation sequencing. It uses the techniques of metagenomics to identify and characterize the genome of bacteria, fungi, parasites, and viruses without the need for a prior knowledge of a specific pathogen directly from clinical specimens. The capacity to detect all the potential pathogens in a sample makes metagenomic next generation sequencing a potent tool in the diagnosis of infectious disease especially when other more directed assays, such as PCR, fail. Its limitations include clinical utility, laboratory validity, sense and sensitivity, cost and regulatory considerations.
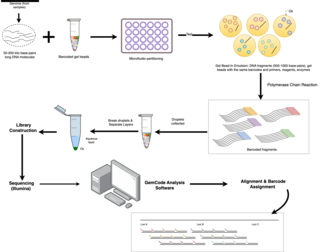
Linked-read sequencing, a type of DNA sequencing technology, uses specialized technique that tags DNA molecules with unique barcodes before fragmenting them. Unlike traditional sequencing technology, where DNA is broken into small fragments and then sequenced individually, resulting in short read lengths that has difficulties in accurately reconstructing the original DNA sequence, the unique barcodes of linked-read sequencing allows scientists to link together DNA fragments that come from the same DNA molecule. A pivotal benefit of this technology lies in the small quantities of DNA required for large genome information output, effectively combining the advantages of long-read and short-read technologies.