The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
Reductive amination is a form of amination that involves the conversion of a carbonyl group to an amine via an intermediate imine. The carbonyl group is most commonly a ketone or an aldehyde. It is a common method to make amines and is widely used in green chemistry since it can be done catalytically in one-pot under mild conditions. In biochemistry, dehydrogenase enzymes use reductive amination to produce the amino acid, glutamate. Additionally, there is ongoing research on alternative synthesis mechanisms with various metal catalysts which allow the reaction to be less energy taxing, and require milder reaction conditions. Investigation into biocatalysts, such as imine reductases, have allowed for higher selectivity in the reduction of chiral amines which is an important factor in pharmaceutical synthesis.
The Carroll rearrangement is a rearrangement reaction in organic chemistry and involves the transformation of a β-keto allyl ester into a α-allyl-β-ketocarboxylic acid. This organic reaction is accompanied by decarboxylation and the final product is a γ,δ-allylketone. The Carroll rearrangement is an adaptation of the Claisen rearrangement and effectively a decarboxylative allylation.

Azomethine ylides are nitrogen-based 1,3-dipoles, consisting of an iminium ion next to a carbanion. They are used in 1,3-dipolar cycloaddition reactions to form five-membered heterocycles, including pyrrolidines and pyrrolines. These reactions are highly stereo- and regioselective, and have the potential to form four new contiguous stereocenters. Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals. Azomethine ylides can be generated from many sources, including aziridines, imines, and iminiums. They are often generated in situ, and immediately reacted with dipolarophiles.

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.

Organocopper chemistry is the study of the physical properties, reactions, and synthesis of organocopper compounds, which are organometallic compounds containing a carbon to copper chemical bond. They are reagents in organic chemistry.
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides. Although Pd-catalyzed C–N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance. The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods while significantly expanding the repertoire of possible C−N bond formations.

Danishefsky's diene is an organosilicon compound and a diene with the formal name trans-1-methoxy-3-trimethylsilyloxy-buta-1,3-diene named after Samuel J. Danishefsky. Because the diene is very electron-rich it is a very reactive reagent in Diels-Alder reactions. This diene reacts rapidly with electrophilic alkenes, such as maleic anhydride. The methoxy group promotes highly regioselective additions. The diene is known to react with amines, aldehydes, alkenes and alkynes. Reactions with imines and nitro-olefins have been reported.
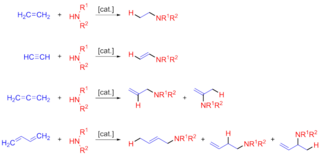
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.

The Shvo catalyst is an organoruthenium compound that catalyzes the hydrogenation of polar functional groups including aldehydes, ketones and imines. The compound is of academic interest as an early example of a catalyst for transfer hydrogenation that operates by an "outer sphere mechanism". Related derivatives are known where p-tolyl replaces some of the phenyl groups. Shvo's catalyst represents a subset of homogeneous hydrogenation catalysts that involves both metal and ligand in its mechanism.

In chemistry, the hydrogenation of carbon–nitrogen double bonds is the addition of the elements of dihydrogen (H2) across a carbon–nitrogen double bond, forming amines or amine derivatives. Although a variety of general methods have been developed for the enantioselective hydrogenation of ketones, methods for the hydrogenation of carbon–nitrogen double bonds are less general. Hydrogenation of imines is complicated by both syn/anti isomerization and tautomerization to enamines, which may be hydrogenated with low enantioselectivity in the presence of a chiral catalyst. Additionally, the substituent attached to nitrogen affects both the reactivity and spatial properties of the imine, complicating the development of a general catalyst system for imine hydrogenation. Despite these challenges, methods have been developed that address particular substrate classes, such as N-aryl, N-alkyl, and endocyclic imines.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
Decarboxylative cross coupling reactions are chemical reactions in which a carboxylic acid is reacted with an organic halide to form a new carbon-carbon bond, concomitant with loss of CO2. Aryl and alkyl halides participate. Metal catalyst, base, and oxidant are required.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. The field is relatively undeveloped compared to research in Lewis acid catalysis.
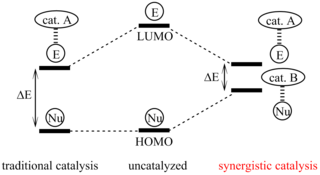
Synergistic catalysis is a specialized approach to catalysis whereby at least two different catalysts act on two different substrates simultaneously to allow reaction between the two activated materials. While a catalyst works to lower the energy of reaction overall, a reaction using synergistic catalysts work together to increase the energy level of HOMO of one of the molecules and lower the LUMO of another. While this concept has come to be important in developing synthetic pathways, this strategy is commonly found in biological systems as well.
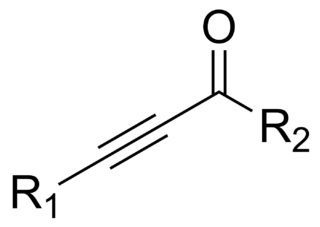
In organic chemistry, an ynone is an organic compound containing a ketone functional group and a C≡C triple bond. Many ynones are α,β-ynones, where the carbonyl and alkyne groups are conjugated. Capillin is a naturally occurring example. Some ynones are not conjugated.
The Crabbé reaction is an organic reaction that converts a terminal alkyne and aldehyde into an allene in the presence of a soft Lewis acid catalyst and secondary amine. Given continued developments in scope and generality, it is a convenient and increasingly important method for the preparation of allenes, a class of compounds often viewed as exotic and synthetically challenging to access.
Chao-Jun "C.-J." Li, a Canadian chemist, is E. B. Eddy Professor of Chemistry and Canada Research Chair in Green Chemistry at McGill University, Montréal. He is known for his pioneering works in Green Solvent and Green Syntheses.

The nitro-Mannich reaction is the nucleophilic addition of a nitroalkane to an imine, resulting in the formation of a beta-nitroamine. With the reaction involving the addition of an acidic carbon nucleophile to a carbon-heteroatom double bond, the nitro-Mannich reaction is related to some of the most fundamental carbon-carbon bond forming reactions in organic chemistry, including the aldol reaction, Henry reaction and Mannich reaction.
The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton. It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.; practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.
