

Baldwin's rules in organic chemistry classify various addition cyclization reactions, and predict when those cyclizations may experience severe steric hindrance during the cyclization step. They were first proposed by Jack Baldwin in 1976. [1] [2]
Baldwin's rules in organic chemistry classify various addition cyclization reactions, and predict when those cyclizations may experience severe steric hindrance during the cyclization step. They were first proposed by Jack Baldwin in 1976. [1] [2]
Baldwin's rules assume that the cyclization is driven by a reactive intermediate, localized to a particular atom. That atom then attacks elsewhere in the molecule (typically, but not necessarily, a multiple bond), breaking one bond and forming a new one. The result is also a less-reactive intermediate localized to one end of the broken bond.
In such cases, three pieces of information suffice to determine geometric constraints on the orbital overlap during the attack:
Baldwin assumed a second-row atom, and so did not address higher coordination numbers. Indeed, Baldwin's rules often fail for rings containing atoms from later periods.[ citation needed ]
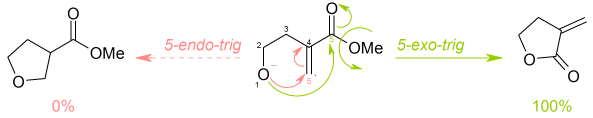
The following ring closure is 5-exo-trig:
In the following tandem reaction, the first step is a 5-exo-dig addition reaction (the second is a Claisen rearrangement): [3]

Gold(I) Lewis acid coordination activates this allene-alkyne for a 6-endo-dig cyclization (and subsequent Nazarov annulation): [4]

The synthesis of (+)-Preussin contains a 5-endo-dig ring closure: [5]

Baldwin discovered that orbital overlap requirements for the formation of bonds favour only certain combinations of ring size and the exo/endo/dig/trig/tet parameters. These rules are based on empirical evidence and exhibit numerous exceptions. [6] [7] [8]
In the case of nucleophilic attack, the requirements follow from the well-understood mechanism geometry. Nucleophiles prefer to attack at 180° (Walden inversion) for exo-tet reactions, 109° (Bürgi–Dunitz angle) for exo-trig reaction, and 120° for endo-dig reactions. [9]
Baldwin's rules also apply to radical intermediates, [10] [11] but cations often disobey Baldwin's rules.[ citation needed ]
Baldwin's original analysis of dig cyclizations has been superseded as inaccurate. The modern rules are as follows: [12]
| 3 | 4 | 5 | 6 | 7 | ||||||
| type | exo | endo | exo | endo | exo | endo | exo | endo | exo | endo |
| tet | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ||
| trig | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ |
| dig | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
Despite the name, these are relative guidelines, not hard-and-fast rules. A disfavoured reaction occurs too slowly to effectively compete with an alternative favoured reaction. However, the disfavoured product may be observed, if no alternate reactions are more favoured.
Baldwin also noted a further constraint on aldol cyclizations. [13] [14] Two new descriptors need to be defined: enolendo and enolexo, which refer to whether both carbons of the enolate C-C fragment are (respectively) incorporated into the ring formed or not. Baldwin found that enolendo cyclizations can only occur in rings with at least 6 members. [15]
