Related Research Articles
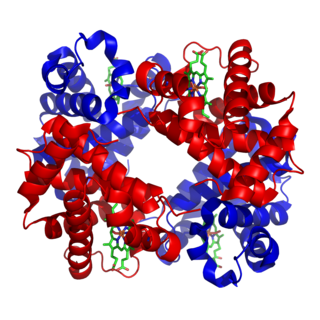
Hemoglobin is a protein containing iron that facilitates the transportation of oxygen in red blood cells. Almost all vertebrates contain hemoglobin, with the sole exception of the fish family Channichthyidae. Hemoglobin in the blood carries oxygen from the respiratory organs to the other tissues of the body, where it releases the oxygen to enable aerobic respiration which powers an animal's metabolism. A healthy human has 12 to 20 grams of hemoglobin in every 100 mL of blood. Hemoglobin is a metalloprotein, a chromoprotein, and globulin.

Hemoglobinopathy is the medical term for a group of inherited blood disorders involving the hemoglobin, the protein of red blood cells. They are single-gene disorders and, in most cases, they are inherited as autosomal co-dominant traits.

Anemia or anaemia is a blood disorder in which the blood has a reduced ability to carry oxygen. This can be due to a lower than normal number of red blood cells, a reduction in the amount of hemoglobin available for oxygen transport, or abnormalities in hemoglobin that impair its function.

Haptoglobin is the protein that in humans is encoded by the HP gene. In blood plasma, haptoglobin binds with high affinity to free hemoglobin released from erythrocytes, and thereby inhibits its deleterious oxidative activity. Compared to Hp, hemopexin binds to free heme. The haptoglobin-hemoglobin complex will then be removed by the reticuloendothelial system.

Thalassemias are inherited blood disorders that manifest as the production of reduced or zero quantities of hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe, including death. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Clinically, thalassemia is classed as Transfusion-Dependent Thalassemia (TDT) or non-Transfusion-Dependent Thalassemia (NTDT), since this determines the principal treatment options. TDT requires regular transfusions, typically every two to five weeks. TDTs include Beta-thalassemia major, non-deletional HbH disease, survived Hb Bart's disease, and severe HbE/beta-thalassemia. NTDT does not need regular transfusions but may require transfusion in case of an anemia crisis.

The Harvard T.H. Chan School of Public Health is the public health school of Harvard University, located in the Longwood Medical Area of Boston, Massachusetts. The school grew out of the Harvard-MIT School for Health Officers, the nation's first graduate training program in population health, which was founded in 1913 and then became the Harvard School of Public Health in 1922.
Hemoglobin A2 (HbA2) is a normal variant of hemoglobin A that consists of two alpha and two delta chains (α2δ2) and is found at low levels in normal human blood. Hemoglobin A2 may be increased in beta thalassemia or in people who are heterozygous for the beta thalassemia gene.
Hemoglobin C is an abnormal hemoglobin in which glutamic acid residue at the 6th position of the β-globin chain is replaced with a lysine residue due to a point mutation in the HBB gene. People with one copy of the gene for hemoglobin C do not experience symptoms, but can pass the abnormal gene on to their children. Those with two copies of the gene are said to have hemoglobin C disease and can experience mild anemia. It is possible for a person to have both the gene for hemoglobin S and the gene for hemoglobin C; this state is called hemoglobin SC disease, and is generally more severe than hemoglobin C disease, but milder than sickle cell anemia.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.
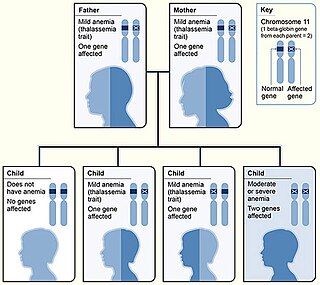
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.
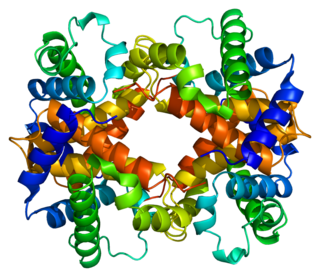
Hemoglobin subunit beta is a globin protein, coded for by the HBB gene, which along with alpha globin (HBA), makes up the most common form of haemoglobin in adult humans, hemoglobin A (HbA). It is 147 amino acids long and has a molecular weight of 15,867 Da. Normal adult human HbA is a heterotetramer consisting of two alpha chains and two beta chains.
William Bosworth Castle was an American physician and physiologist who transformed hematology from a "descriptive art to a dynamic interdisciplinary science."
Yuet Wai Kan, is a Chinese-American geneticist and hematologist. He is the current Louis K. Diamond Chair in Hematology and a Professor Emeritus at the University of California, San Francisco. He is a former president of the American Society of Hematology.

Hemoglobin E (HbE) is an abnormal hemoglobin with a single point mutation in the β chain. At position 26 there is a change in the amino acid, from glutamic acid to lysine (E26K). Hemoglobin E is very common among people of Southeast Asian, Northeast Indian, Sri Lankan and Bangladeshi descent.

Sickle cell disease (SCD), also simply called sickle cell, is a group of hemoglobin-related blood disorders that are typically inherited. The most common type is known as sickle cell anemia. Sickle cell anemia results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells. This leads to the red blood cells adopting an abnormal sickle-like shape under certain circumstances; with this shape, they are unable to deform as they pass through capillaries, causing blockages. Problems in sickle cell disease typically begin around 5 to 6 months of age. A number of health problems may develop, such as attacks of pain in joints, anemia, swelling in the hands and feet, bacterial infections, dizziness and stroke. The probability of severe symptoms, including long-term pain, increases with age. Without treatment, people with SCD rarely reach adulthood but with good healthcare, median life expectancy is between 58 and 66 years. All the major organs are affected by sickle cell disease. The liver, heart, kidneys, gallbladder, eyes, bones, and joints also can suffer damage from the abnormal functions of the sickle cells, and their inability to flow through the small blood vessels correctly.

Michael R. Hayden, is a Killam Professor of Medical Genetics at the University of British Columbia, the highest honour UBC can confer on any faculty member. Only four such awards have ever been conferred in the Faculty of Medicine. Hayden is best known for his research in Huntington disease (HD).
Stuart Holland Orkin is an American physician, stem cell biologist and researcher in pediatric hematology-oncology. He is the David G. Nathan Distinguished Professor of Pediatrics at Harvard Medical School. Orkin's research has focused on the genetic basis of blood disorders. He is a member of the National Academy of Sciences and the Institute of Medicine, and an Investigator of the Howard Hughes Medical Institute.
BioViva is an American biotechnology gene therapy company, based in Bainbridge Island, Washington, researching treatments to interfere in the aging process in humans.

Virginia Minnich (1910–1996) was an American molecular biologist and hematology researcher known for discovering hemoglobin E, an abnormal form of hemoglobin that can cause blood disorders, and for working out the glutathione synthesis pathway. She was a noted blood morphologist and teacher and helped set up hematology laboratories around the world. She was the first person without a PhD or MD to be appointed a Professor of Medicine at Washington University School of Medicine.
References
- ↑ "Dr. Barry Paw Service Details". 2017. Archived from the original on 12 January 2018. Retrieved 11 January 2018.
- ↑ "Physician Profile: Barry H. Paw, M.D." Board of Registration in Medicine. Commonwealth of Massachusetts. 2012. Archived from the original on 4 March 2016. Retrieved 8 April 2012.
- ↑ "Burmese Researcher Helps Discover New Gene". ဗွီအိုအေ (in Burmese). 2006-03-05. Archived from the original on 2023-05-22. Retrieved 2023-05-22.
- ↑ "Barry H. Paw". Biological and Biomedical Sciences. Harvard College. 2012. Archived from the original on 24 June 2014. Retrieved 8 April 2012.
- ↑ Kaplan, Jerry (January 3, 2018). "In Memoriam In Memory of Barry Paw". International Society for the Study of Iron in Biology and Medicine. Archived from the original on May 14, 2024. Retrieved May 14, 2024.
| | This article about a Burmese scientist is a stub. You can help Wikipedia by expanding it. |