The following outline is provided as an overview of and topical guide to organic chemistry:
In chemistry, stereoselectivity is the property of a chemical reaction in which a single reactant forms an unequal mixture of stereoisomers during a non-stereospecific creation of a new stereocenter or during a non-stereospecific transformation of a pre-existing one. The selectivity arises from differences in steric and electronic effects in the mechanistic pathways leading to the different products. Stereoselectivity can vary in degree but it can never be total since the activation energy difference between the two pathways is finite: both products are at least possible and merely differ in amount. However, in favorable cases, the minor stereoisomer may not be detectable by the analytic methods used.
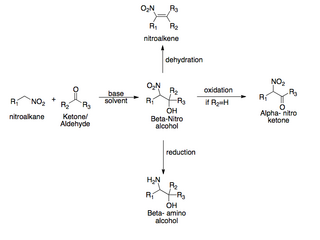
The Henry reaction is a classic carbon–carbon bond formation reaction in organic chemistry. Discovered in 1895 by the Belgian chemist Louis Henry (1834–1913), it is the combination of a nitroalkane and an aldehyde or ketone in the presence of a base to form β-nitro alcohols. This type of reaction is also referred to as a nitroaldol reaction. It is nearly analogous to the aldol reaction that had been discovered 23 years prior that couples two carbonyl compounds to form β-hydroxy carbonyl compounds known as "aldols". The Henry reaction is a useful technique in the area of organic chemistry due to the synthetic utility of its corresponding products, as they can be easily converted to other useful synthetic intermediates. These conversions include subsequent dehydration to yield nitroalkenes, oxidation of the secondary alcohol to yield α-nitro ketones, or reduction of the nitro group to yield β-amino alcohols.

A nucleophilic aromatic substitution (SNAr) is a substitution reaction in organic chemistry in which the nucleophile displaces a good leaving group, such as a halide, on an aromatic ring. Aromatic rings are usually nucleophilic, but some aromatic compounds do undergo nucleophilic substitution. Just as normally nucleophilic alkenes can be made to undergo conjugate substitution if they carry electron-withdrawing substituents, so normally nucleophilic aromatic rings also become electrophilic if they have the right substituents.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.
A pinacol coupling reaction is an organic reaction in which a carbon–carbon bond is formed between the carbonyl groups of an aldehyde or a ketone in presence of an electron donor in a free radical process. The reaction product is a vicinal diol. The reaction is named after pinacol, which is the product of this reaction when done with acetone as reagent. The reaction is usually a homocoupling but intramolecular cross-coupling reactions are also possible. Pinacol was discovered by Wilhelm Rudolph Fittig in 1859.

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.

Asymmetric induction describes the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral feature present in the substrate, reagent, catalyst or environment. Asymmetric induction is a key element in asymmetric synthesis.
The Wulff–Dötz reaction (also known as the Dötz reaction or the benzannulation reaction of the Fischer carbene complexes) is the chemical reaction of an aromatic or vinylic alkoxy pentacarbonyl chromium carbene complex with an alkyne and carbon monoxide to give a Cr(CO)3-coordinated substituted phenol. Several reviews have been published. It is named after the German chemist Karl Heinz Dötz (b. 1943) and the American chemist William D. Wulff (b. 1949) at Michigan State University. The reaction was first discovered by Karl Dötz and was extensively developed by his group and W. Wulff's group. They subsequently share the name of the reaction.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
(Benzene)chromium tricarbonyl is an organometallic compound with the formula Cr(C6H6)(CO)3. This yellow crystalline solid compound is soluble in common nonpolar organic solvents. The molecule adopts a geometry known as “piano stool” because of the planar arrangement of the aryl group and the presence of three CO ligands as "legs" on the chromium-bond axis.
Reductions with hydrosilanes are methods used for hydrogenation and hydrogenolysis of organic compounds. The approach is a subset of ionic hydrogenation. In this particular method, the substrate is treated with a hydrosilane and auxiliary reagent, often a strong acid, resulting in formal transfer of hydride from silicon to carbon. This style of reduction with hydrosilanes enjoys diverse if specialized applications.
Heteroatom-promoted lateral lithiation is the site-selective replacement of a benzylic hydrogen atom for lithium for the purpose of further functionalization. Heteroatom-containing substituents may direct metalation to the benzylic site closest to the heteroatom or increase the acidity of the ring carbons via an inductive effect.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
In chemistry, metal-catalysed hydroboration is a reaction used in organic synthesis. It is one of several examples of homogeneous catalysis.
Electrophilic aromatic substitution (SEAr) is an organic reaction in which an atom that is attached to an aromatic system is replaced by an electrophile. Some of the most important electrophilic aromatic substitutions are aromatic nitration, aromatic halogenation, aromatic sulfonation, alkylation Friedel–Crafts reaction and acylation Friedel–Crafts reaction.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. The field is relatively undeveloped compared to research in Lewis acid catalysis.
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.

Ugi’s amine is an organometallic compound with the formula (C5H5)Fe(C5H4CH N 2. It is named for the chemist who first reported its synthesis in 1970, Ivar Ugi. It is a ferrocene derivative. Ugi’s amine is a precursor to ligands, most notably, the Josiphos ligands, which have been used in asymmetric catalysis










