Prader–Willi syndrome (PWS) is a genetic disorder caused by a loss of function of specific genes on chromosome 15. In newborns, symptoms include weak muscles, poor feeding, and slow development. Beginning in childhood, those affected become constantly hungry, which often leads to obesity and type 2 diabetes. Mild to moderate intellectual impairment and behavioral problems are also typical of the disorder. Often, affected individuals have a narrow forehead, small hands and feet, short height, and light skin and hair. Most are unable to have children.

Kabuki syndrome is a congenital disorder of genetic origin. It affects multiple parts of the body, with varying symptoms and severity, although the most common is the characteristic facial appearance.

Coffin–Lowry syndrome is a genetic disorder that is X-linked dominant and which causes severe mental problems sometimes associated with abnormalities of growth, cardiac abnormalities, kyphoscoliosis, as well as auditory and visual abnormalities.

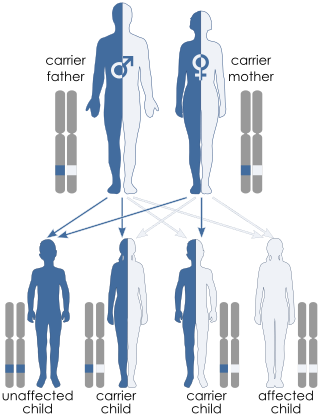
Jacobsen syndrome is a rare chromosomal disorder resulting from deletion of genes from chromosome 11 that includes band 11q24.1. It is a congenital disorder. Since the deletion takes place on the q arm of chromosome 11, it is also called 11q terminal deletion disorder. The deletion may range from 5 million to 16 million deleted DNA base pairs. The severity of symptoms depends on the number of deletions; the more deletions there are, the more severe the symptoms are likely to be.
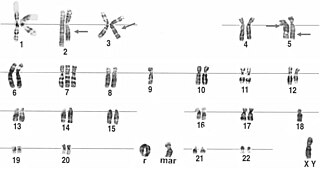
A ring chromosome is an aberrant chromosome whose ends have fused together to form a ring. Ring chromosomes were first discovered by Lilian Vaughan Morgan in 1926. A ring chromosome is denoted by the symbol r in human genetics and R in Drosophila genetics. Ring chromosomes may form in cells following genetic damage by mutagens like radiation, but they may also arise spontaneously during development.

Hyperlysinemia is an autosomal recessive metabolic disorder characterized by an abnormal increase of lysine in the blood, but appears to be benign. It is caused by mutations in AASS, which encodes α-aminoadipic semialdehyde synthase.

Barakat syndrome is a rare disease characterized by hypoparathyroidism, sensorineural deafness and renal disease, and hence also known as HDR syndrome. It was first described by Amin J. Barakat et al. in 1977.
Abruzzo–Erickson syndrome is an extremely rare disorder characterized by deafness, protruding ears, coloboma, a cleft palate or palatal rugosity, radial synostosis, and short stature. It was first characterized by Abruzzo and Erickson in 1977 as a CHARGE like syndrome as variably expressed among a family of two brothers, their mother, and their maternal uncle. Members of this family exhibited many of the CHARGE symptoms, but notably did not have choanal atresia and the brothers experienced typical genital development. Due to the recent discovery of this disorder, its etiology is not fully known but it is understood that it arises from mutations on the TBX22 gene on the X-chromosome. The disorder is inherited in an X-linked recessive manner. There is currently no known cure but its symptoms can be treated.
Naegeli–Franceschetti–Jadassohn syndrome (NFJS), also known as chromatophore nevus of Naegeli and Naegeli syndrome, is a rare autosomal dominant form of ectodermal dysplasia, characterized by reticular skin pigmentation, diminished function of the sweat glands, the absence of teeth and hyperkeratosis of the palms and soles. One of the most striking features is the absence of fingerprint lines on the fingers.
Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.
Björnstad syndrome is an autosomal recessive congenital condition involving pili torti, sensorineural deafness, and hair abnormalities. It was first characterized in 1965, in Oslo, by prof. Roar Theodor Bjørnstad after he observed an association between pili torti and hearing loss. The condition is extremely rare, with less than 50 cases documented in medical literature worldwide.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
9q34 deletion syndrome is a rare genetic disorder. Terminal deletions of chromosome 9q34 have been associated with childhood hypotonia, a distinctive facial appearance and developmental disability. The facial features typically described include arched eyebrows, small head circumference, midface hypoplasia, prominent jaw and a pouting lower lip. Individuals with this disease may often have speech impediments, such as speech delays. Other characteristics of this disease include: epilepsy, congenital and urogenital defects, microcephaly, corpulence, and psychiatric disorders. From analysis of chromosomal breakpoints, as well as gene sequencing in suggestive cases, Kleefstra and colleagues identified EHMT1 as the causative gene. This gene is responsible for producing the protein histone methyltransferase which functions to alter histones. Ultimately, histone methyltransferases are important in deactivating certain genes, needed for proper growth and development. Moreover, a frameshift, missense, or nonsense error in the coding sequence of EHMT1 can result in this condition in an individual.

Ohad Birk, a physician-scientist, is a professor of human genetics, converging basic scientific research with effective clinical translational applications. Birk's research lab deciphered the molecular basis and mechanism of more than 30 human diseases, including some of the most prevalent severe hereditary diseases in Arabs and in Jews, as well as three syndromes named after Birk. He also implemented his scientific findings in massive carrier testing programs, conducive to 30% reduction in infant mortality rate in the Bedouin community, as well as near-eradication of two of the most common severe hereditary diseases in Sephardic Jews. Birk heads the clinical Genetics Institute at Soroka Medical Center and the Morris Kahn Laboratory of Human Genetics as well as Israel's National Research Center for Orphan / Rare Diseases at Ben Gurion University, and served as director of Israel's National Institute of Biotechnology in the Negev (NIBN) between 2016 and 2017.

Arts syndrome is a rare metabolic disorder that causes serious neurological problems in males due to a malfunction of the PRPP synthetase 1 enzyme. Arts Syndrome is part of a spectrum of PRPS-1 related disorders with reduced activity of the enzyme that includes Charcot–Marie–Tooth disease and X-linked non-syndromic sensorineural deafness.
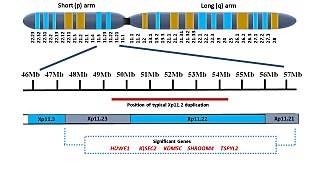
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).
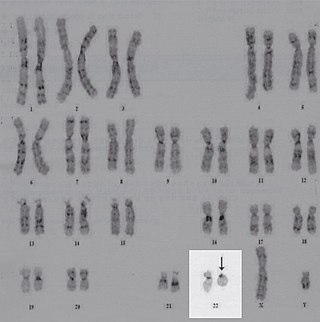
Ring chromosome 22, also known as ring 22, is a rare chromosomal disorder. Ring chromosomes occur when the ends of a chromosome lose material and fuse into a ring shape; in the case of ring 22, this occurs for chromosome 22, the last numbered human autosome. Ring chromosome 22 is marked by a number of consistent traits, such as intellectual disability, speech delay, hypotonia, and hyperactivity. The condition has a similar phenotype to Phelan-McDermid syndrome, as the loss of the SHANK3 gene is implicated in both.
Leigh syndrome, French Canadian type, also known as congenital lactic acidosis, Saguenay-Lac-Saint-Jean type, is a rare mitochondrial disorder which is characterized by regular metabolic acidosis, hypotonia, developmental delays and facial dysmorphy. It's associated with mutations in a gene in chromosome 2. Approximately 100 cases of this syndrome have been reported in medical literature.
Multiple congenital anomalies-hypotonia-seizures syndrome is a rare multi-systemic genetic disorder which is characterized by developmental delay, seizures, hypotonia and heart, urinary, and gastrointestinal abnormalities.
CHAMP1-associated intellectual disability syndrome, also known as autosomal dominant intellectual disability type 40, is a rare genetic disorder characterized by intellectual disabilities, developmental delays, facial dysmorphisms, and other anomalies.