
Aniridia is the absence of the iris, a muscular structure that opens and closes the pupil to allow light into the eye. It is also responsible for eye color. Without it the central eye appears all black. It can be congenital, in which both eyes are usually involved, or caused by a penetrant injury. Isolated aniridia is a congenital disorder which is not limited to a defect in iris development, but is a panocular condition with macular and optic nerve hypoplasia, cataract, and corneal changes. Vision may be severely compromised and the disorder is frequently associated with a number of ocular complications: nystagmus, amblyopia, buphthalmos, and cataract. Aniridia in some individuals occurs as part of a syndrome, such as WAGR syndrome, or Gillespie syndrome.
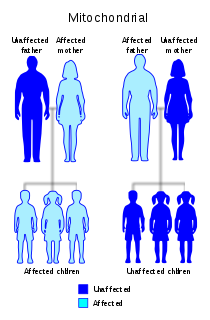
Leber's hereditary optic neuropathy (LHON) is a mitochondrially inherited degeneration of retinal ganglion cells (RGCs) and their axons that leads to an acute or subacute loss of central vision; it predominantly affects young adult males. LHON is transmitted only through the mother, as it is primarily due to mutations in the mitochondrial genome, and only the egg contributes mitochondria to the embryo. LHON is usually due to one of three pathogenic mitochondrial DNA (mtDNA) point mutations. These mutations are at nucleotide positions 11778 G to A, 3460 G to A and 14484 T to C, respectively in the ND4, ND1 and ND6 subunit genes of complex I of the oxidative phosphorylation chain in mitochondria. Men cannot pass on the disease to their offspring.

Walker–Warburg syndrome (WWS), also called Warburg syndrome, Chemke syndrome, HARD syndrome, Pagon syndrome, cerebroocular dysgenesis (COD) or cerebroocular dysplasia-muscular dystrophy syndrome (COD-MD), is a rare form of autosomal recessive congenital muscular dystrophy. It is associated with brain and eye abnormalities. This condition has a worldwide distribution. The overall incidence is unknown but a survey in North-eastern Italy has reported an incidence rate of 1.2 per 100,000 live births. It is the most severe form of congenital muscular dystrophy with most children dying before the age of three years.
Dominant optic atrophy, or dominant optic atrophy, Kjer's type, is an autosomally inherited disease that affects the optic nerves, causing reduced visual acuity and blindness beginning in childhood. This condition is due to mitochondrial dysfunction mediating the death of optic nerve fibers. Dominant optic atrophy was first described clinically by Batten in 1896 and named Kjer’s optic neuropathy in 1959 after Danish ophthalmologist Poul Kjer, who studied 19 families with the disease. Although dominant optic atrophy is the most common autosomally inherited optic neuropathy aside from glaucoma, it is often misdiagnosed.

GAPO syndrome is a rare, autosomal recessive disorder that causes severe growth retardation, and has been observed fewer than 30 times before 2011. GAPO is an acronym that encompasses the predominant traits of the disorder: growth retardation, alopecia, pseudoanodontia, and worsening optic atrophy in some subjects. Other common symptoms include premature aging, large, prominent foreheads, and delayed bone aging. GAPO syndrome typically results in premature death around age 30–40, due to interstitial fibrosis and atherosclerosis.
Costeff syndrome, or 3-methylglutaconic aciduria type III, is a genetic disorder caused by mutations in the OPA3 gene. It is typically associated with the onset of visual deterioration in early childhood followed by the development of movement problems and motor disability in later childhood, occasionally along with mild cases of cognitive deficiency. The disorder is named after Hanan Costeff, the doctor who first described the syndrome in 1989.

Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
9q34 deletion syndrome is a rare genetic disorder. Terminal deletions of chromosome 9q34 have been associated with childhood hypotonia, a distinctive facial appearance and developmental disability. The facial features typically described include arched eyebrows, small head circumference, midface hypoplasia, prominent jaw and a pouting lower lip. Individuals with this disease may often have speech impediments, such as speech delays. Other characteristics of this disease include: epilepsy, congenital and urogenital defects, microcephaly, corpulence, and psychiatric disorders. From analysis of chromosomal breakpoints, as well as gene sequencing in suggestive cases, Kleefstra and colleagues identified EHMT1 as the causative gene. This gene is responsible for producing the protein histone methyltransferase which functions to alter histones. Ultimately, histone methyltransferases are important in deactivating certain genes, needed for proper growth and development. Moreover, a frameshift, missense, or nonsense error in the coding sequence of EHMT1 can result in this condition in an individual.

Arts syndrome is a rare metabolic disorder that causes serious neurological problems in males due to a malfunction of the PRPP synthetase 1 enzyme. Arts Syndrome is part of a spectrum of PRPS-1 related disorders with reduced activity of the enzyme that includes Charcot–Marie–Tooth disease and X-linked non-syndromic sensorineural deafness.
ZTTK syndrome is a rare disease caused in humans by a genetic mutation of the SON gene. Common symptoms include developmental delay and sometimes moderate to several intellectual disability.
Birk-Barel syndrome is a rare genetic disorder associated with the KCNK9 gene. Signs and symptoms include mental retardation, hypotonia, hyperactivity, and syndromic facies.
Kenny-Caffey syndrome type 2 (KCS2) is an extremely rare autosomal dominant genetic condition characterized by dwarfism, hypermetropia, microphthalmia, and skeletal abnormalities. This subtype of Kenny-Caffey syndrome is caused by a heterozygous mutation in the FAM111A gene (615292) on chromosome 11q12.
Bainbridge–Ropers syndrome is a very rare genetic disorder characterized by abnormalities including severe psychomotor development, feeding problems, severe postnatal growth delays, intellectual disabilities, and skeletal abnormalities.
ZC4H2 is a protein-coding gene located on the X-chromosome. This gene encodes a protein which is a member of the so-called zinc finger domain-containing protein family. There is currently very limited understanding about the ZC4H2 gene and its protein function.

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.
DDX3X syndrome is a genetic disorder that affects predominantly females. Patients with DDX3X syndrome may develop developmental delay or intellectual disability, autism, ADHD, and low muscle tone. The syndrome develops due to mutations of the DDX3X gene located on the X chromosome, and the clinical picture varies depending on the specific mutation.
PURA syndrome, also known as PURA-related neurodevelopmental disorder, is a rare novel genetic disorder which is characterized by developmental and speech delay, neo-natal hypotonia, failure to thrive, excessive sleepiness, epilepsy, and other anomalies.
Proud syndrome is a very rare genetic disorder which is characterized by severe intellectual disabilities, corpus callosum agenesis, epilepsy, and spasticity. It is a type of syndromic X-linked intellectual disability.
Goldmann-Favre syndrome is a rare genetic disorder characterized by early-onset nyctalopia, decreased visual acuity, and abnormal findings of the fundus. It is a type of progressive vitreotapetoretinal degeneration.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
