Related Research Articles
In molecular biology, restriction fragment length polymorphism (RFLP) is a technique that exploits variations in homologous DNA sequences, known as polymorphisms, populations, or species or to pinpoint the locations of genes within a sequence. The term may refer to a polymorphism itself, as detected through the differing locations of restriction enzyme sites, or to a related laboratory technique by which such differences can be illustrated. In RFLP analysis, a DNA sample is digested into fragments by one or more restriction enzymes, and the resulting restriction fragments are then separated by gel electrophoresis according to their size.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.

In genetics, dominance is the phenomenon of one variant (allele) of a gene on a chromosome masking or overriding the effect of a different variant of the same gene on the other copy of the chromosome. The first variant is termed dominant and the second is called recessive. This state of having two different variants of the same gene on each chromosome is originally caused by a mutation in one of the genes, either new or inherited. The terms autosomal dominant or autosomal recessive are used to describe gene variants on non-sex chromosomes (autosomes) and their associated traits, while those on sex chromosomes (allosomes) are termed X-linked dominant, X-linked recessive or Y-linked; these have an inheritance and presentation pattern that depends on the sex of both the parent and the child. Since there is only one copy of the Y chromosome, Y-linked traits cannot be dominant or recessive. Additionally, there are other forms of dominance, such as incomplete dominance, in which a gene variant has a partial effect compared to when it is present on both chromosomes, and co-dominance, in which different variants on each chromosome both show their associated traits.
Genetic linkage is the tendency of DNA sequences that are close together on a chromosome to be inherited together during the meiosis phase of sexual reproduction. Two genetic markers that are physically near to each other are unlikely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be more linked than markers that are far apart. In other words, the nearer two genes are on a chromosome, the lower the chance of recombination between them, and the more likely they are to be inherited together. Markers on different chromosomes are perfectly unlinked, although the penetrance of potentially deleterious alleles may be influenced by the presence of other alleles, and these other alleles may be located on other chromosomes than that on which a particular potentially deleterious allele is located.
A genetic screen or mutagenesis screen is an experimental technique used to identify and select individuals who possess a phenotype of interest in a mutagenized population. Hence a genetic screen is a type of phenotypic screen. Genetic screens can provide important information on gene function as well as the molecular events that underlie a biological process or pathway. While genome projects have identified an extensive inventory of genes in many different organisms, genetic screens can provide valuable insight as to how those genes function.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.
A quantitative trait locus (QTL) is a locus that correlates with variation of a quantitative trait in the phenotype of a population of organisms. QTLs are mapped by identifying which molecular markers correlate with an observed trait. This is often an early step in identifying the actual genes that cause the trait variation.
Forward genetics is a molecular genetics approach of determining the genetic basis responsible for a phenotype. Forward genetics provides an unbiased approach because it relies heavily on identifying the genes or genetic factors that cause a particular phenotype or trait of interest.
A genetic marker is a gene or DNA sequence with a known location on a chromosome that can be used to identify individuals or species. It can be described as a variation that can be observed. A genetic marker may be a short DNA sequence, such as a sequence surrounding a single base-pair change, or a long one, like minisatellites.

Random amplified polymorphic DNA (RAPD), pronounced "rapid", is a type of polymerase chain reaction (PCR), but the segments of DNA that are amplified are random. The scientist performing RAPD creates several arbitrary, short primers, then proceeds with the PCR using a large template of genomic DNA, hoping that fragments will amplify. By resolving the resulting patterns, a semi-unique profile can be gleaned from an RAPD reaction.
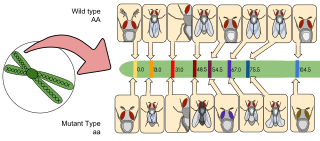
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.
Genetic association is when one or more genotypes within a population co-occur with a phenotypic trait more often than would be expected by chance occurrence.
Genotyping is the process of determining differences in the genetic make-up (genotype) of an individual by examining the individual's DNA sequence using biological assays and comparing it to another individual's sequence or a reference sequence. It reveals the alleles an individual has inherited from their parents. Traditionally genotyping is the use of DNA sequences to define biological populations by use of molecular tools. It does not usually involve defining the genes of an individual.
A molecular marker is a molecule, sampled from some source, that gives information about its source. For example, DNA is a molecular marker that gives information about the organism from which it was taken. For another example, some proteins can be molecular markers of Alzheimer's disease in a person from which they are taken. Molecular markers may be non-biological. Non-biological markers are often used in environmental studies.
Marker assisted selection or marker aided selection (MAS) is an indirect selection process where a trait of interest is selected based on a marker linked to a trait of interest, rather than on the trait itself. This process has been extensively researched and proposed for plant- and animal- breeding.
In genetics, association mapping, also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes to genotypes, uncovering genetic associations.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.

Combined Bisulfite Restriction Analysis is a molecular biology technique that allows for the sensitive quantification of DNA methylation levels at a specific genomic locus on a DNA sequence in a small sample of genomic DNA. The technique is a variation of bisulfite sequencing, and combines bisulfite conversion based polymerase chain reaction with restriction digestion. Originally developed to reliably handle minute amounts of genomic DNA from microdissected paraffin-embedded tissue samples, the technique has since seen widespread usage in cancer research and epigenetics studies.
Quantitative trait loci mapping or QTL mapping is the process of identifying genomic regions that potentially contain genes responsible for important economic, health or environmental characters. Mapping QTLs is an important activity that plant breeders and geneticists routinely use to associate potential causal genes with phenotypes of interest. Family-based QTL mapping is a variant of QTL mapping where multiple-families are used.
A human disease modifier gene is a modifier gene that alters expression of a human gene at another locus that in turn causes a genetic disease. Whereas medical genetics has tended to distinguish between monogenic traits, governed by simple, Mendelian inheritance, and quantitative traits, with cumulative, multifactorial causes, increasing evidence suggests that human diseases exist on a continuous spectrum between the two.
References
- ↑ McClean, Phillip (1992). "Specialized Mapping Topics". North Dakota State University. Retrieved 9 November 2014.
- ↑ Henke, K; Bowen, M; Harris, M (August 15, 2013). "Perspectives for identification of mutations in the zebrafish: Making use of next-generation sequencing technologies for forward genetic approaches". Methods. 62 (3): 185–196. doi:10.1016/j.ymeth.2013.05.015. PMID 23748111.
- ↑ Michelmore, R; Paran, I; Kesseli, R (November 1, 1991). "Identification of Markers Linked to Disease-Resistance Genes by Bulked Segregant Analysis: A Rapid Method to Detect Markers in Specific Genomic Regions by Using Segregating Populations". Proceedings of the National Academy of Sciences of the United States of America. 88 (21): 9828–9832. doi: 10.1073/pnas.88.21.9828 . PMC 52814 . PMID 1682921.