Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.

DNA synthesis is the natural or artificial creation of deoxyribonucleic acid (DNA) molecules. DNA is a macromolecule made up of nucleotide units, which are linked by covalent bonds and hydrogen bonds, in a repeating structure. DNA synthesis occurs when these nucleotide units are joined to form DNA; this can occur artificially or naturally. Nucleotide units are made up of a nitrogenous base, pentose sugar (deoxyribose) and phosphate group. Each unit is joined when a covalent bond forms between its phosphate group and the pentose sugar of the next nucleotide, forming a sugar-phosphate backbone. DNA is a complementary, double stranded structure as specific base pairing occurs naturally when hydrogen bonds form between the nucleotide bases.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional mutating changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.

In molecular biology, a library is a collection of DNA fragments that is stored and propagated in a population of micro-organisms through the process of molecular cloning. There are different types of DNA libraries, including cDNA libraries, genomic libraries and randomized mutant libraries. DNA library technology is a mainstay of current molecular biology, genetic engineering, and protein engineering, and the applications of these libraries depend on the source of the original DNA fragments. There are differences in the cloning vectors and techniques used in library preparation, but in general each DNA fragment is uniquely inserted into a cloning vector and the pool of recombinant DNA molecules is then transferred into a population of bacteria or yeast such that each organism contains on average one construct. As the population of organisms is grown in culture, the DNA molecules contained within them are copied and propagated.
A DNA construct is an artificially-designed segment of DNA borne on a vector that can be used to incorporate genetic material into a target tissue or cell. A DNA construct contains a DNA insert, called a transgene, delivered via a transformation vector which allows the insert sequence to be replicated and/or expressed in the target cell. This gene can be cloned from a naturally occurring gene, or synthetically constructed. The vector can be delivered using physical, chemical or viral methods. Typically, the vectors used in DNA constructs contain an origin of replication, a multiple cloning site, and a selectable marker. Certain vectors can carry additional regulatory elements based on the expression system involved.
The overlap extension polymerase chain reaction is a variant of PCR. It is also referred to as Splicing by overlap extension / Splicing by overhang extension (SOE) PCR. It is used assemble multiple smaller double stranded DNA fragments into a larger DNA sequence. OE-PCR is widely used to insert mutations at specific points in a sequence or to assemble custom DNA sequence from smaller DNA fragments into a larger polynucleotide.
Recombineering is a genetic and molecular biology technique based on homologous recombination systems, as opposed to the older/more common method of using restriction enzymes and ligases to combine DNA sequences in a specified order. Recombineering is widely used for bacterial genetics, in the generation of target vectors for making a conditional mouse knockout, and for modifying DNA of any source often contained on a bacterial artificial chromosome (BAC), among other applications.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
An allele-specific oligonucleotide (ASO) is a short piece of synthetic DNA complementary to the sequence of a variable target DNA. It acts as a probe for the presence of the target in a Southern blot assay or, more commonly, in the simpler dot blot assay. It is a common tool used in genetic testing, forensics, and molecular biology research.
Artificial gene synthesis, or simply gene synthesis, refers to a group of methods that are used in synthetic biology to construct and assemble genes from nucleotides de novo. Unlike DNA synthesis in living cells, artificial gene synthesis does not require template DNA, allowing virtually any DNA sequence to be synthesized in the laboratory. It comprises two main steps, the first of which is solid-phase DNA synthesis, sometimes known as DNA printing. This produces oligonucleotide fragments that are generally under 200 base pairs. The second step then involves connecting these oligonucleotide fragments using various DNA assembly methods. Because artificial gene synthesis does not require template DNA, it is theoretically possible to make a completely synthetic DNA molecule with no limits on the nucleotide sequence or size.
The versatility of polymerase chain reaction (PCR) has led to modifications of the basic protocol being used in a large number of variant techniques designed for various purposes. This article summarizes many of the most common variations currently or formerly used in molecular biology laboratories; familiarity with the fundamental premise by which PCR works and corresponding terms and concepts is necessary for understanding these variant techniques.
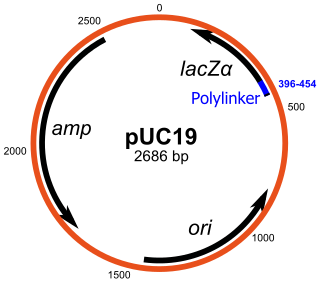
pUC19 is one of a series of plasmid cloning vectors created by Joachim Messing and co-workers. The designation "pUC" is derived from the classical "p" prefix and the abbreviation for the University of California, where early work on the plasmid series had been conducted. It is a circular double stranded DNA and has 2686 base pairs. pUC19 is one of the most widely used vector molecules as the recombinants, or the cells into which foreign DNA has been introduced, can be easily distinguished from the non-recombinants based on color differences of colonies on growth media. pUC18 is similar to pUC19, but the MCS region is reversed.
The Gateway cloning method, invented and commercialized by Invitrogen since the late 1990s, is the cloning method of the integration and excision recombination reactions that take place when bacteriophage lambda infects bacteria. This technology provides a fast and highly efficient way to transport DNA sequences into multi-vector systems for functional analysis and protein expression using Gateway att sites, and two proprietary enzyme mixes called BP Clonase and LR Clonase. In vivo, these recombination reactions are facilitated by the recombination of attachment sites from the lambda/phage chromosome (attP) and the bacteria (attB). As a result of recombination between the attP and attB sites, the phage integrates into the bacterial genome flanked by two new recombination sites. The removal of the phage from the bacterial chromosome and the regeneration of attP and attB sites can both result from the attL and attR sites recombining under specific circumstances.
Delitto perfetto is a genetic technique for in vivo site-directed mutagenesis in yeast. This name is the Italian term for "perfect murder", and it refers to the ability of the technique to create desired genetic changes without leaving any foreign DNA in the genome.

Genetic engineering techniques allow the modification of animal and plant genomes. Techniques have been devised to insert, delete, and modify DNA at multiple levels, ranging from a specific base pair in a specific gene to entire genes. There are a number of steps that are followed before a genetically modified organism (GMO) is created. Genetic engineers must first choose what gene they wish to insert, modify, or delete. The gene must then be isolated and incorporated, along with other genetic elements, into a suitable vector. This vector is then used to insert the gene into the host genome, creating a transgenic or edited organism.

In molecular biology, mutagenesis is an important laboratory technique whereby DNA mutations are deliberately engineered to produce libraries of mutant genes, proteins, strains of bacteria, or other genetically modified organisms. The various constituents of a gene, as well as its regulatory elements and its gene products, may be mutated so that the functioning of a genetic locus, process, or product can be examined in detail. The mutation may produce mutant proteins with interesting properties or enhanced or novel functions that may be of commercial use. Mutant strains may also be produced that have practical application or allow the molecular basis of a particular cell function to be investigated.

Golden Gate Cloning or Golden Gate assembly is a molecular cloning method that allows a researcher to simultaneously and directionally assemble multiple DNA fragments into a single piece using Type IIS restriction enzymes and T4 DNA ligase. This assembly is performed in vitro. Most commonly used Type IIS enzymes include BsaI, BsmBI, and BbsI.

Duplex sequencing is a library preparation and analysis method for next-generation sequencing (NGS) platforms that employs random tagging of double-stranded DNA to detect mutations with higher accuracy and lower error rates.
No-SCAR genome editing is an editing method that is able to manipulate the Escherichia coli genome. The system relies on recombineering whereby DNA sequences are combined and manipulated through homologous recombination. No-SCAR is able to manipulate the E. coli genome without the use of the chromosomal markers detailed in previous recombineering methods. Instead, the λ-Red recombination system facilitates donor DNA integration while Cas9 cleaves double-stranded DNA to counter-select against wild-type cells. Although λ-Red and Cas9 genome editing are widely used technologies, the no-SCAR method is novel in combining the two functions; this technique is able to establish point mutations, gene deletions, and short sequence insertions in several genomic loci with increased efficiency and time sensitivity.
This glossary of cellular and molecular biology is a list of definitions of terms and concepts commonly used in the study of cell biology, molecular biology, and related disciplines, including genetics, biochemistry, and microbiology. It is split across two articles:
