
The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.

The heritability of autism is the proportion of differences in expression of autism that can be explained by genetic variation; if the heritability of a condition is high, then the condition is considered to be primarily genetic. Autism has a strong genetic basis. Although the genetics of autism are complex, autism spectrum disorder (ASD) is explained more by multigene effects than by rare mutations with large effects.

Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.

The causes of autism are environmental or genetic factors that predispose an individual to develop autism, also known as autism spectrum disorder (ASD). Many causes of autism have been proposed, but understanding of the theory of causation of autism is incomplete. Attempts have been made to incorporate the known genetic and environmental causes into a comprehensive causative framework. ASD is a neurodevelopmental disorder marked by impairments in communicative ability and social interaction and restricted/repetitive behaviors, interests, or activities not suitable for the individual's developmental stage. The severity of symptoms and functional impairment vary between individuals.

Human genetic variation is the genetic differences in and among populations. There may be multiple variants of any given gene in the human population (alleles), a situation called polymorphism.
The Olduvai domain, known until 2018 as DUF1220 and the NBPF repeat, is a protein domain that shows a striking human lineage-specific (HLS) increase in copy number and appears to be involved in human brain evolution. The protein domain has also been linked to several neurogenetic disorders such as schizophrenia and increased severity of autism. In 2018, it was named by its discoverers after Olduvai Gorge in Tanzania, one of the most important archaeological sites for early humans, to reflect data indicating its role in human brain size and evolution.

In genomics, a genome-wide association study, is an observational study of a genome-wide set of genetic variants in different individuals to see if any variant is associated with a trait. GWA studies typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major human diseases, but can equally be applied to any other genetic variants and any other organisms.

The 1000 Genomes Project, launched in January 2008, was an international research effort to establish by far the most detailed catalogue of human genetic variation. Scientists planned to sequence the genomes of at least one thousand anonymous participants from a number of different ethnic groups within the following three years, using newly developed technologies which were faster and less expensive. In 2010, the project finished its pilot phase, which was described in detail in a publication in the journal Nature. In 2012, the sequencing of 1092 genomes was announced in a Nature publication. In 2015, two papers in Nature reported results and the completion of the project and opportunities for future research.
The Centre for Applied Genomics is a genome centre in the Research Institute of The Hospital for Sick Children, and is affiliated with the University of Toronto. TCAG also operates as a Science and Technology Innovation Centre of Genome Canada, with an emphasis on next-generation sequencing (NGS) and bioinformatics support. Research at TCAG focuses on the genetic and genomic basis of human variability, health and disease, including research on the genetics of autism spectrum disorder and structural variation of the human genome. The centre is located in the Peter Gilgan Centre for Research and Learning in downtown Toronto, Canada.
Complete Genomics is a life sciences company that has developed and commercialized a DNA sequencing platform for human genome sequencing and analysis. This solution combines the company's proprietary human genome sequencing technology with its informatics and data management software to provide finished variant reports and assemblies at Complete Genomics’ own commercial genome center in Mountain View, California.
Expression quantitative trait loci (eQTLs) are genomic loci that explain variation in expression levels of mRNAs.
esophageal candidiasis1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.

Stephen Wayne "Steve" Scherer is a Canadian scientist who studies genetic variation in human disease. He obtained his PhD at the University of Toronto under Professor Lap-chee Tsui. Together they founded Canada's first human genome centre, the Centre for Applied Genomics (TCAG) at the Hospital for Sick Children. He continues to serve as director of TCAG, and is also a University Professor in the Department of Molecular Genetics, and the director of the McLaughlin Centre at the Temerty Faculty of Medicine at the University of Toronto.
Genomic structural variation is the variation in structure of an organism's chromosome. It consists of many kinds of variation in the genome of one species, and usually includes microscopic and submicroscopic types, such as deletions, duplications, copy-number variants, insertions, inversions and translocations. Originally, a structure variation affects a sequence length about 1kb to 3Mb, which is larger than SNPs and smaller than chromosome abnormality. However, the operational range of structural variants has widened to include events > 50bp. The definition of structural variation does not imply anything about frequency or phenotypical effects. Many structural variants are associated with genetic diseases, however many are not. Recent research about SVs indicates that SVs are more difficult to detect than SNPs. Approximately 13% of the human genome is defined as structurally variant in the normal population, and there are at least 240 genes that exist as homozygous deletion polymorphisms in human populations, suggesting these genes are dispensable in humans. Rapidly accumulating evidence indicates that structural variations can comprise millions of nucleotides of heterogeneity within every genome, and are likely to make an important contribution to human diversity and disease susceptibility.

Eric Emil Schadt is an American mathematician and computational biologist. He is founder and former chief executive officer of Sema4, a patient-centered health intelligence company, and dean for precision medicine and Mount Sinai Professor in Predictive Health and Computational Biology at the Icahn School of Medicine at Mount Sinai. He was previously founding director of the Icahn Institute for Genomics and Multiscale Biology and chair of the Department of Genetics and Genomics Sciences at the Icahn School of Medicine at Mount Sinai.
Predictive genomics is at the intersection of multiple disciplines: predictive medicine, personal genomics and translational bioinformatics. Specifically, predictive genomics deals with the future phenotypic outcomes via prediction in areas such as complex multifactorial diseases in humans. To date, the success of predictive genomics has been dependent on the genetic framework underlying these applications, typically explored in genome-wide association (GWA) studies. The identification of associated single-nucleotide polymorphisms underpin GWA studies in complex diseases that have ranged from Type 2 Diabetes (T2D), Age-related macular degeneration (AMD) and Crohn's disease.
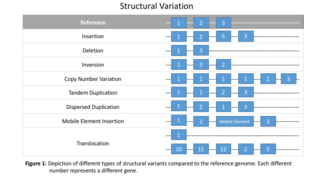
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.

Complex traits, also known as quantitative traits, are traits that do not behave according to simple Mendelian inheritance laws. More specifically, their inheritance cannot be explained by the genetic segregation of a single gene. Such traits show a continuous range of variation and are influenced by both environmental and genetic factors. Compared to strictly Mendelian traits, complex traits are far more common, and because they can be hugely polygenic, they are studied using statistical techniques such as quantitative genetics and quantitative trait loci (QTL) mapping rather than classical genetics methods. Examples of complex traits include height, circadian rhythms, enzyme kinetics, and many diseases including diabetes and Parkinson's disease. One major goal of genetic research today is to better understand the molecular mechanisms through which genetic variants act to influence complex traits.
The GWAS catalog is a free online database that compiles data of genome-wide association studies (GWAS), summarizing unstructured data from different literature sources into accessible high quality data. It was created by the National Human Genome Research Institute (NHGRI) in 2008 and have become a collaborative project between the NHGRI and the European Bioinformatics Institute (EBI) since 2010. As of September 2018, it has included 71,673 SNP–trait associations in 3,567 publications.

Andre Franke, born on 16 October 1978, is a geneticist, academic, and university professor. He is a Full W3 Professor of Molecular Medicine at the Christian-Albrechts-University of Kiel, and a managing director at the Institute of Clinical Molecular Biology.
