
Good manufacturing practices (GMP) are the practices required in order to conform to the guidelines recommended by agencies that control the authorization and licensing of the manufacture and sale of food and beverages, cosmetics, pharmaceutical products, dietary supplements, and medical devices. These guidelines provide minimum requirements that a manufacturer must meet to assure that their products are consistently high in quality, from batch to batch, for their intended use. The rules that govern each industry may differ significantly; however, the main purpose of GMP is always to prevent harm from occurring to the end user. Additional tenets include ensuring the end product is free from contamination, that it is consistent in its manufacture, that its manufacture has been well documented, that personnel are well trained, and that the product has been checked for quality more than just at the end phase. GMP is typically ensured through the effective use of a quality management system (QMS).

CE marking is an administrative marking with which the manufacturer or importer affirms its conformity with European health, safety, and environmental protection standards for products sold within the European Economic Area (EEA). It is not a quality indicator or a certification mark. The CE marking is also found on products sold outside the EEA that have been manufactured to EEA standards. This makes the CE marking recognizable worldwide even to people who are not familiar with the European Economic Area. It is in that sense like the FCC Declaration of Conformity used for selling certain electronic devices in the United States.

A medical device is any device intended to be used for medical purposes. Medical devices benefit patients by helping health care providers diagnose and treat patients and helping patients overcome sickness or disease, improving their quality of life. Significant potential for hazards are inherent when using a device for medical purposes and thus medical devices must be proved safe and effective with reasonable assurance before regulating governments allow marketing of the device in their country. As a general rule, as the associated risk of the device increases the amount of testing required to establish safety and efficacy also increases. Further, as associated risk increases the potential benefit to the patient must also increase.
Regulatory affairs (RA), also called government affairs, is a profession within regulated industries, such as pharmaceuticals, medical devices, cosmetics, agrochemicals, energy, banking, telecom etc. Regulatory affairs also has a very specific meaning within the healthcare industries.
Type approval or certificate of conformity is granted to a product that meets a minimum set of regulatory, technical and safety requirements. Generally, type approval is required before a product is allowed to be sold in a particular country, so the requirements for a given product will vary around the world. Processes and certifications known as type approval in English are generally called homologation, or some cognate expression, in other European languages.
ISO 13485Medical devices -- Quality management systems -- Requirements for regulatory purposes is an International Organization for Standardization (ISO) standard published for the first time in 1996; it represents the requirements for a comprehensive quality management system for the design and manufacture of medical devices. This standard supersedes earlier documents such as EN 46001 and EN 46002 (1996), the previously published ISO 13485, and ISO 13488.
Postmarketing surveillance (PMS), also known as post market surveillance, is the practice of monitoring the safety of a pharmaceutical drug or medical device after it has been released on the market and is an important part of the science of pharmacovigilance. Since drugs and medical devices are approved on the basis of clinical trials, which involve relatively small numbers of people who have been selected for this purpose – meaning that they normally do not have other medical conditions which may exist in the general population – postmarketing surveillance can further refine, or confirm or deny, the safety of a drug or device after it is used in the general population by large numbers of people who have a wide variety of medical conditions.
A European Authorised Representative (E.A.R.) serves as a legal entity designated by non European Union (EU) manufacturers, to represent them in the EU and ensure their compliance with the European Directives.
The Revised Payment Services Directive is an EU Directive, administered by the European Commission to regulate payment services and payment service providers throughout the European Union (EU) and European Economic Area (EEA). The PSD's purpose was to increase pan-European competition and participation in the payments industry also from non-banks, and to provide for a level playing field by harmonizing consumer protection and the rights and obligations for payment providers and users. The key objectives of the PSD2 directive are creating a more integrated European payments market, making payments more secure and protecting consumers.
Design controls designates the application of a formal methodology to the conduct of product development activities. It is often mandatory to implement such practice when designing and developing products within regulated industries.
A notified body, in the European Union, is an organisation that has been designated by a member state to assess the conformity of certain products, before being placed on the EU market, with the applicable essential technical requirements. These essential requirements are publicised in European directives or regulations.
The Unique Device Identification (UDI) System is intended to assign a unique identifier to medical devices within the United States, Europe, China, South Korea, Saudi Arabia and Taiwan. It was signed into law in the US on September 27, 2007, as part of the Food and Drug Administration Amendments Act of 2007. The EU acted to adopt UDI and on April 5, 2017, under the EU Medical Device Regulation (MDR) and In-vitro Diagnostic Regulation (IVDR), but adoption has been postponed to 2021; see Medical Device Regulation.
ISO 14971Medical devices — Application of risk management to medical devices is an ISO standard for the application of risk management to medical devices. The ISO Technical Committee responsible for the maintenance of this standard is ISO TC 210 working with IEC/SC62A through Joint Working Group one (JWG1). This standard is the culmination of the work starting in ISO/IEC Guide 51, and ISO/IEC Guide 63. The latest significant revision was published in 2019. In 2013, a technical report ISO/TR 24971 was published by ISO TC 210 to provide expert guidance on the application of this standard.
The international standard IEC 62304 – medical device software – software life cycle processes is a standard which specifies life cycle requirements for the development of medical software and software within medical devices. It is harmonized by the European Union (EU) and the United States (US), and therefore can be used as a benchmark to comply with regulatory requirements from both these markets.
In 2009, a European Commission initiative resulted in the specification of a common external power supply for use with data-enabled mobile phones sold in the European Union. The external power supply is the AC electric power adapter that converts household AC electricity voltages to the much lower DC voltages needed to charge a mobile phone's internal battery. Although compliance is voluntary, a majority of the world's largest mobile phone manufacturers agreed to make their applicable mobile phones compatible with Europe's common external power supply specification.

Regulation No. 305/2011 of the European Parliament and of the European Council is a regulation of 9 March 2011 that lays down harmonised conditions for the marketing of construction products and replaces Construction Products Directive (89/106/EEC). The EU regulation is designed to simplify and clarify the existing framework for the placing on the market of construction products.
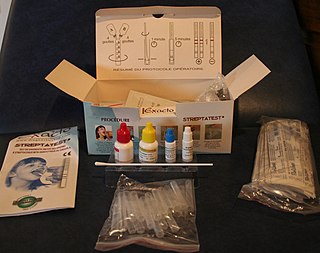
A rapid diagnostic test (RDT) is a medical diagnostic test that is quick and easy to perform. RDTs are suitable for preliminary or emergency medical screening and for use in medical facilities with limited resources. They also allow point-of-care testing in primary care for things that formerly only a laboratory test could measure. They provide same-day results within two hours, typically in approximately 20 minutes.
A technical file is a set of documents that describes a product and can prove that the product was designed and according to the requirements of a quality management system.
Regulation (EU) 2017/745 is a regulation of the European Union on the clinical investigation and sale of medical devices for human use. It repeals Directive 93/42/EEC, which concerns medical devices, and Directive 90/385/EEC, which concerns active implantable medical devices, on 26 May 2021.
Regulation (EU) 2017/746 (IVDR) is a regulation of the European Union on the placing on the market and putting into service of in vitro diagnostic medical devices (IVD), repealing Directive 98/79/EC (IVDD), which also concerned IVD. The regulation was published in April 2017 and is closely aligned to the EU regulation on medical devices. Changes compared to the IVDD include changes in device classification, stricter oversight of manufacturers by Notified Bodies, introduction of the "Person Responsible for Regulatory Compliance" (PRRC), the requirement of UDI marking for devices, common specifications, Eudamed registration, and increased post-market surveillance activities.