
Heritability is a statistic used in the fields of breeding and genetics that estimates the degree of variation in a phenotypic trait in a population that is due to genetic variation between individuals in that population. The concept of heritability can be expressed in the form of the following question: "What is the proportion of the variation in a given trait within a population that is not explained by the environment or random chance?"
Twin studies are studies conducted on identical or fraternal twins. They aim to reveal the importance of environmental and genetic influences for traits, phenotypes, and disorders. Twin research is considered a key tool in behavioral genetics and in related fields, from biology to psychology. Twin studies are part of the broader methodology used in behavior genetics, which uses all data that are genetically informative – siblings studies, adoption studies, pedigree, etc. These studies have been used to track traits ranging from personal behavior to the presentation of severe mental illnesses such as schizophrenia.
Genetic linkage is the tendency of DNA sequences that are close together on a chromosome to be inherited together during the meiosis phase of sexual reproduction. Two genetic markers that are physically near to each other are unlikely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be more linked than markers that are far apart. In other words, the nearer two genes are on a chromosome, the lower the chance of recombination between them, and the more likely they are to be inherited together. Markers on different chromosomes are perfectly unlinked, although the penetrance of potentially deleterious alleles may be influenced by the presence of other alleles, and these other alleles may be located on other chromosomes than that on which a particular potentially deleterious allele is located.
A quantitative trait locus (QTL) is a locus that correlates with variation of a quantitative trait in the phenotype of a population of organisms. QTLs are mapped by identifying which molecular markers correlate with an observed trait. This is often an early step in identifying the actual genes that cause the trait variation.
Human genetics is the study of inheritance as it occurs in human beings. Human genetics encompasses a variety of overlapping fields including: classical genetics, cytogenetics, molecular genetics, biochemical genetics, genomics, population genetics, developmental genetics, clinical genetics, and genetic counseling.
Genetics, a discipline of biology, is the science of heredity and variation in living organisms.
The candidate gene approach to conducting genetic association studies focuses on associations between genetic variation within pre-specified genes of interest, and phenotypes or disease states. This is in contrast to genome-wide association studies (GWAS), which is a hypothesis-free approach that scans the entire genome for associations between common genetic variants and traits of interest. Candidate genes are most often selected for study based on a priori knowledge of the gene's biological functional impact on the trait or disease in question. The rationale behind focusing on allelic variation in specific, biologically relevant regions of the genome is that certain alleles within a gene may directly impact the function of the gene in question and lead to variation in the phenotype or disease state being investigated. This approach often uses the case-control study design to try to answer the question, "Is one allele of a candidate gene more frequently seen in subjects with the disease than in subjects without the disease?" Candidate genes hypothesized to be associated with complex traits have generally not been replicated by subsequent GWASs or highly powered replication attempts. The failure of candidate gene studies to shed light on the specific genes underlying such traits has been ascribed to insufficient statistical power, low prior probability that scientists can correctly guess a specific allele within a specific gene that is related to a trait, poor methodological practices, and data dredging.
A polygene is a member of a group of non-epistatic genes that interact additively to influence a phenotypic trait, thus contributing to multiple-gene inheritance, a type of non-Mendelian inheritance, as opposed to single-gene inheritance, which is the core notion of Mendelian inheritance. The term "monozygous" is usually used to refer to a hypothetical gene as it is often difficult to distinguish the effect of an individual gene from the effects of other genes and the environment on a particular phenotype. Advances in statistical methodology and high throughput sequencing are, however, allowing researchers to locate candidate genes for the trait. In the case that such a gene is identified, it is referred to as a quantitative trait locus (QTL). These genes are generally pleiotropic as well. The genes that contribute to type 2 diabetes are thought to be mostly polygenes. In July 2016, scientists reported identifying a set of 355 genes from the last universal common ancestor (LUCA) of all organisms living on Earth.
Genetic association is when one or more genotypes within a population co-occur with a phenotypic trait more often than would be expected by chance occurrence.
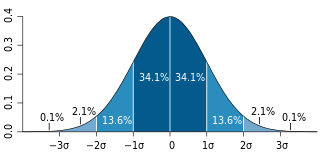
In mathematical or statistical modeling a threshold model is any model where a threshold value, or set of threshold values, is used to distinguish ranges of values where the behaviour predicted by the model varies in some important way. A particularly important instance arises in toxicology, where the model for the effect of a drug may be that there is zero effect for a dose below a critical or threshold value, while an effect of some significance exists above that value. Certain types of regression model may include threshold effects.
Genetic epidemiology is the study of the role of genetic factors in determining health and disease in families and in populations, and the interplay of such genetic factors with environmental factors. Genetic epidemiology seeks to derive a statistical and quantitative analysis of how genetics work in large groups.
In multivariate quantitative genetics, a genetic correlation is the proportion of variance that two traits share due to genetic causes, the correlation between the genetic influences on a trait and the genetic influences on a different trait estimating the degree of pleiotropy or causal overlap. A genetic correlation of 0 implies that the genetic effects on one trait are independent of the other, while a correlation of 1 implies that all of the genetic influences on the two traits are identical. The bivariate genetic correlation can be generalized to inferring genetic latent variable factors across > 2 traits using factor analysis. Genetic correlation models were introduced into behavioral genetics in the 1970s–1980s.

Behavioural genetics, also referred to as behaviour genetics, is a field of scientific research that uses genetic methods to investigate the nature and origins of individual differences in behaviour. While the name "behavioural genetics" connotes a focus on genetic influences, the field broadly investigates the extent to which genetic and environmental factors influence individual differences, and the development of research designs that can remove the confounding of genes and environment. Behavioural genetics was founded as a scientific discipline by Francis Galton in the late 19th century, only to be discredited through association with eugenics movements before and during World War II. In the latter half of the 20th century, the field saw renewed prominence with research on inheritance of behaviour and mental illness in humans, as well as research on genetically informative model organisms through selective breeding and crosses. In the late 20th and early 21st centuries, technological advances in molecular genetics made it possible to measure and modify the genome directly. This led to major advances in model organism research and in human studies, leading to new scientific discoveries.
Quantitative trait loci mapping or QTL mapping is the process of identifying genomic regions that potentially contain genes responsible for important economic, health or environmental characters. Mapping QTLs is an important activity that plant breeders and geneticists routinely use to associate potential causal genes with phenotypes of interest. Family-based QTL mapping is a variant of QTL mapping where multiple-families are used.
Oligogenic inheritance describes a trait that is influenced by a few genes. Oligogenic inheritance represents an intermediate between monogenic inheritance in which a trait is determined by a single causative gene, and polygenic inheritance, in which a trait is influenced by many genes and often environmental factors.

Genetic variance is a concept outlined by the English biologist and statistician Ronald Fisher in his fundamental theorem of natural selection. In his 1930 book The Genetical Theory of Natural Selection, Fisher postulates that the rate of change of biological fitness can be calculated by the genetic variance of the fitness itself. Fisher tried to give a statistical formula about how the change of fitness in a population can be attributed to changes in the allele frequency. Fisher made no restrictive assumptions in his formula concerning fitness parameters, mate choices or the number of alleles and loci involved.
Genome-wide complex trait analysis (GCTA) Genome-based restricted maximum likelihood (GREML) is a statistical method for variance component estimation in genetics which quantifies the total narrow-sense (additive) contribution to a trait's heritability of a particular subset of genetic variants. This is done by directly quantifying the chance genetic similarity of unrelated individuals and comparing it to their measured similarity on a trait; if two unrelated individuals are relatively similar genetically and also have similar trait measurements, then the measured genetics are likely to causally influence that trait, and the correlation can to some degree tell how much. This can be illustrated by plotting the squared pairwise trait differences between individuals against their estimated degree of relatedness. The GCTA framework can be applied in a variety of settings. For example, it can be used to examine changes in heritability over aging and development. It can also be extended to analyse bivariate genetic correlations between traits. There is an ongoing debate about whether GCTA generates reliable or stable estimates of heritability when used on current SNP data. The method is based on the outdated and false dichotomy of genes versus the environment. It also suffers from serious methodological weaknesses, such as susceptibility to population stratification.
A human disease modifier gene is a modifier gene that alters expression of a human gene at another locus that in turn causes a genetic disease. Whereas medical genetics has tended to distinguish between monogenic traits, governed by simple, Mendelian inheritance, and quantitative traits, with cumulative, multifactorial causes, increasing evidence suggests that human diseases exist on a continuous spectrum between the two.

Family resemblance refers to physical similarities shared between close relatives, especially between parents and children and between siblings. In psychology, the similarities of personality are also observed.

Complex traits, also known as quantitative traits, are traits that do not behave according to simple Mendelian inheritance laws. More specifically, their inheritance cannot be explained by the genetic segregation of a single gene. Such traits show a continuous range of variation and are influenced by both environmental and genetic factors. Compared to strictly Mendelian traits, complex traits are far more common, and because they can be hugely polygenic, they are studied using statistical techniques such as quantitative genetics and quantitative trait loci (QTL) mapping rather than classical genetics methods. Examples of complex traits include height, circadian rhythms, enzyme kinetics, and many diseases including diabetes and Parkinson's disease. One major goal of genetic research today is to better understand the molecular mechanisms through which genetic variants act to influence complex traits.