| Craniorhiny | |
|---|---|
 | |
| Autosomal dominant inheritance |
Craniorhiny is a rare autosomal dominant syndrome characterized by craniosynostosis (oxycephaly) and facial anomalies around the nose base and lips. [1] [2]
| Craniorhiny | |
|---|---|
| | |
| Autosomal dominant inheritance |
Craniorhiny is a rare autosomal dominant syndrome characterized by craniosynostosis (oxycephaly) and facial anomalies around the nose base and lips. [1] [2]
Features of this condition include: [1] [3]
Infranasal spherical cyst-like formations with fistulas have also been seen. [3]
The first (and only confirmed) reports of this condition was made in 1991, seen in a father and son. Two siblings reported in 2007 are also speculated to have the condition. [2]

A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Turricephaly is a type of cephalic disorder where the head appears tall with a small length and width. It is due to premature closure of the coronal suture plus any other suture, like the lambdoid, or it may be used to describe the premature fusion of all sutures. It should be differentiated from Crouzon syndrome. Oxycephaly is a form of turricephaly where the head is cone-shaped, and is the most severe of the craniosynostoses.

Waardenburg syndrome is a group of rare genetic conditions characterised by at least some degree of congenital hearing loss and pigmentation deficiencies, which can include bright blue eyes, a white forelock or patches of light skin. These basic features constitute type 2 of the condition; in type 1, there is also a wider gap between the inner corners of the eyes called telecanthus, or dystopia canthorum. In type 3, which is rare, the arms and hands are also malformed, with permanent finger contractures or fused fingers, while in type 4, the person also has Hirschsprung's disease. There also exist at least two types that can result in central nervous system (CNS) symptoms such as developmental delay and muscle tone abnormalities.

Mismatch repair cancer syndrome (MMRCS) is a cancer syndrome associated with biallelic DNA mismatch repair mutations. It is also known as Turcot syndrome and by several other names.

Craniosynostosis is a condition in which one or more of the fibrous sutures in a young infant's skull prematurely fuses by turning into bone (ossification), thereby changing the growth pattern of the skull. Because the skull cannot expand perpendicular to the fused suture, it compensates by growing more in the direction parallel to the closed sutures. Sometimes the resulting growth pattern provides the necessary space for the growing brain, but results in an abnormal head shape and abnormal facial features. In cases in which the compensation does not effectively provide enough space for the growing brain, craniosynostosis results in increased intracranial pressure leading possibly to visual impairment, sleeping impairment, eating difficulties, or an impairment of mental development combined with a significant reduction in IQ.

Hypochondroplasia (HCH) is a developmental disorder caused by an autosomal dominant genetic defect in the fibroblast growth factor receptor 3 gene (FGFR3) that results in a disproportionately short stature, micromelia and a head that appears large in comparison with the underdeveloped portions of the body. It is classified as short-limbed dwarfism.
Acromicric dysplasia is an extremely rare inherited disorder characterized by abnormally short hands and feet, growth retardation and delayed bone maturation leading to short stature. Most cases have occurred randomly for no apparent reason (sporadically). However, autosomal dominant inheritance has not been ruled out.
Fazio–Londe disease (FLD), also called progressive bulbar palsy of childhood, is a very rare inherited motor neuron disease of children and young adults and is characterized by progressive paralysis of muscles innervated by cranial nerves.
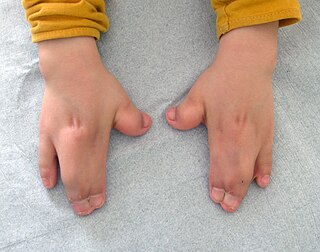
Acrocephalosyndactyly is a group of autosomal dominant congenital disorders characterized by craniofacial (craniosynostosis) and hand and foot (syndactyly) abnormalities. When polydactyly is present, the classification is acrocephalopolysyndactyly. Acrocephalosyndactyly is mainly diagnosed postnatally, although prenatal diagnosis is possible if the mutation is known to be within the family genome. Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.
Raine syndrome (RNS), also called osteosclerotic bone dysplasia, is a rare autosomal recessive congenital disorder characterized by craniofacial anomalies including microcephaly, noticeably low set ears, osteosclerosis, a cleft palate, gum hyperplasia, a hypoplastic nose, and eye proptosis. It is considered to be a lethal disease, and usually leads to death within a few hours of birth. However, a recent report describes two studies in which children with Raine syndrome have lived to 8 and 11 years old, so it is currently proposed that there is a milder expression that the phenotype can take.

Hanhart syndrome is a broadly classified medical condition consisting of congenital disorders that cause an undeveloped tongue and malformed extremities and fingers. There exist five types of Hanhart syndrome, with the severity and nature of the condition ranging widely on a case-by-case basis. Hanhart syndrome is classified as a rare disease, with approximately 30 known cases having been reported between 1932 and 1991. Early hypotheses believed that the disorder was caused by genetic conditions, with a more recent hypothesis demonstrating evidence that the disorder may be caused by hemorrhagic lesions during prenatal development. The causal mechanism behind this vascular disruption is still unknown.
Sanjad-Sakati syndrome is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. It was first described in Saudi Arabia, but has been seen in Qatari, Kuwaiti, Omani and other children from the Middle East as well as elsewhere. The condition is caused by mutations or deletions in the TBCE gene of Chromosome No.1.
Acromesomelic dysplasia is a rare skeletal disorder that causes abnormal bone and cartilage development, leading to shortening of the forearms, lower legs, hands, feet, fingers, and toes. Five different genetic mutations have been implicated in the disorder. Treatment is individualized but is generally aimed at palliating symptoms, for example, treatment of kyphosis and lumbar hyperlordosis.

A bifid nose is an uncommon congenital malformation which is characterized by the presence of a cleft between the two nostrils of the nose. It is the result of a disturbance during embryological nose development.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
Pyknoachondrogenesis is a very rare, fatal, presumably autosomal recessive genetic disorder characterized by symptoms similar to those shown by patients with achondrogenesis alongside severely osteoclerotic bones and early death. The findings that can be seen in patients with this condition include hydrops fetalis, palpebral edemas, low-set ears, abdomen prominence, short neck, large head, depressed nasal bridge, shortening and widening of the trunk, severe short-limbed dwarfism, craniofacial hyperostosis, agenesis of the pubic bones, hypoplasia of the pelvic bones and ischium, poor ossification of the vertebrae and sacrum, webbing of the neck, and shortening of a long bone and the ribs. Pregnancies of babies with this condition generally aren't compatible with life and they end up in miscarriage, stillbirth, or in neonatal death. Only 5 cases from Italy and the United States, respectively, have been described in medical literature.
Ventricular extrasystoles with syncopal episodes-perodactyly-Robin sequence syndrome is a rare autosomal dominant genetic disorder characterized by cardiofaciodigital anomalies occurring alongside Pierre Robin sequence. Additional features include abnormal sense of smell, camptodactyly, recurrent joint dislocations, and short stature. Around 6 to 12 cases have been described in medical literature.

Craniosynostosis and dental anomalies is an autosomal recessive syndrome characterized by craniosynostosis, maxillary hypoplasia, and dental anomalies. Dental anomalies seen in this condition include malocclusion, delayed and ectopic tooth eruption, and/or supernumerary teeth. Syndactyly, clinodactyly, and other digit anomalies may also be present.
Craniosynostosis, Adelaide type (CRSA) is a syndrome characterized by cone-shaped epiphyses, phalangeal hypoplasia, and carpal bone malsegmentation along with craniosynostosis.

Craniosynostosis-Dandy-Walker malformation-hydrocephalus syndrome is an autosomal dominant syndrome characterized by sagittal craniosynostosis (scaphocephaly), Dandy-Walker malformation, hydrocephalus, and craniofacial dysmorphisms including hypertelorism, micrognathia, and positional ear deformities.