Cycloisomerization is any isomerization in which the cyclicisomer of the substrate is produced in the reaction coordinate. The greatest advantage of cycloisomerization reactions is its atom economical nature, by design nothing is wasted, as every atom in the starting material is present in the product. In most cases these reactions are mediated by a transition metal catalyst, in few cases organocatalysts and rarely do they occur under thermal conditions. These cyclizations are able to be performed with excellent levels of selectivity in numerous cases and have transformed cycloisomerization into a powerful tool for unique and complex molecular construction.[1] Cycloisomerization is a very broad topic in organic synthesis and many reactions that would be categorized as such exist. Two basic classes of these reactions are intramolecularMichael addition and Intramolecular Diels–Alder reactions. Under the umbrella of cycloisomerization, enyne and related olefin cycloisomerizations are the most widely used and studied reactions.[2]
A rather intuitive route to cyclic isomers is the intramolecular conjugate addition to α,β–unsaturated carbonyls (intramolecular Michael addition or IMA). Competent Michael acceptors include conjugated enones, enals or nitroalkene derivatives and examples of other acceptors are sparse.[3] Despite IMA reactions being ubiquitous in synthesis, very few examples of asymmetric IMA transformations exist.[2]
Thiourea catalysts with pendant chiral backbones have shown to activate systems with tethered nitroalkane and ester motifs to induce asymmetric IMA.[3][4] The utility of this transformation was demonstrated in the synthesis of cyclic γ– amino acid precursors (figure 1).[3] It is proposed that activation occurs via H–bonding of both the nitronate and the ester to the thiourea catalyst and explains the interesting selectivity for the E–ester.[3]
A functional stereodivergent organocatalyzed IMA/lactonization transformation in the synthesis of substituted dihydrofurans and tetrahydrofurans has been studied for its ability to construct important structural motifs in numerous natural products (figure 2).[5] When ethers such as 3 are subject to (S)–(–)–tetramisole hydrochloride (4) catalyst the result is the syn–2,3–substituted THF while the complementary anti–product is easily accessible via a Cinchona alkaloid catalyst such as 7.[5]
Intramolecular Diels–Alder
Intramolecular Diels–Alder (IMDA) reactions pair tethered dienes and dienophiles in a [4+2] fashion, the most common being terminal substitution. These transformations are popular in total synthesis and have seen a wide spread use in advance to numerous difficult synthetic targets.[6] One such use is the application of an enantioselective IMDA transformation in the asymmetric synthesis of the marine toxin (–)–isopulo'upone (10).[7]
The synthesis of (–)–isopulo'upone demonstrated the utility of cationic Cu(II)bis(oxazoline) complex catalyzed IMDA reactions to give bicyclic products with as many as four neighboring stereogenic centers (figure 3).[7] A rather recent application of IMDA reactions in complex molecule synthesis is the IMDA approach to the tricyclic core of palhinine lycopodium alkaloids, a class of natural products isolated from nodding club moss.[8]
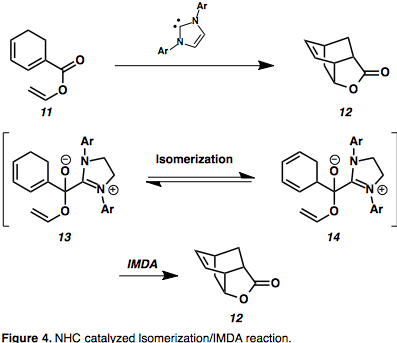
N–heterocyclic carbenes (NHCs) are an emerging class of organocatalysts that are able to induce Umpolung reactivity as well as normal polarity transformations, however until recently these have not been broadly used in total synthesis due to limited substrate scope.[9] An interesting expansion in the use of these organocatalysts is the NHC catalyzed olefin isomerization/IMDA cascade reaction to give unique bicyclic scaffolds.[10][11] Dienyl esters such as 11 were transformed into substituted bicyclo[2.2.2]octanes via an isomerization step stabilized by a hemiacetal azolium intermediate (13).[11] The activation barrier of isomerization of 1,3–hexadiene through a [1,5]–shift is 41 Kcal mol–1 and is expected to increase with conjugation to the ester, thus uncatalyzed isomerization is unlikely.[11] This provides the advantage of bypassing a high barrier of activation, providing access to previously unobtainable IMDA derivatives.
Enyne cycloisomerization
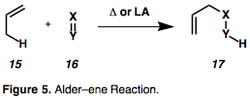
Enyne cycloisomerization, an alkyne variant of the Alder-ene reaction (figure 5), is an intramolecular rearrangement of 1,n–enynes to give the corresponding cyclic isomer.
Although the rearrangement may occur under thermal conditions, the scope of the thermal rearrangement is limited due to the requirement of high temperatures, thus transition metals such as Au, Pd, Pt, Rh and Ir are often employed as catalysts.[2] As synthesis quarrels to build complex structural motifs in the presence of inductive, stereoelectronic and steric demands this rearrangement has recently been developed as a robust method for constructing carbo– and heterocyclic scaffolds with excellent chemo–, regio– and diastereoselective outcomes.[2]
There is not a single mechanism that can be used to describe enyne cycloisomerizations as the mechanism depends on reaction conditions and catalyst selection.[2][12] Intermediates of the metal catalyzed cycloisomerization in which the metal coordinates the alkyne or alkene activating either or both are possible and are shown in figure 6.
Activation of the alkyne by complexation with the metal leading to an η2–metal intermediate such as 18 opens up the alkyne to nucleophilic attack and engenders carbocation intermediates. This Pull–push reactivity is important for understanding reactions mediated by π–acids. Complexation of the alkyne to the metal fragment depletes electron density in the bond (‘pull’), in concert with the ability of the metal to back donate (“push”) arouses the observed consecutive electrophilic and nucleophilic character to the vicinal carbon atoms of the alkyne (figure 7).
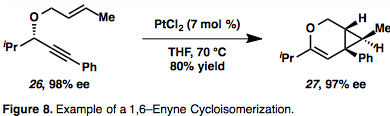
Metallacycle intermediates (19) are the result of the simultaneous complexation and activation of both partners. Hydrometallation of the alkyne giving a vinyl metal species that may in turn carbometalate the olefin is also possible (20). An example of a 1,6–enyne cycloisomerization proceeding through an η2–activated metal intermediate is given in figure 8,[13] which is common for enyne cycloisomerizations mediated by Pt or Au due to their π–acidic nature. Notably, in this example chirality transfer takes place in which the absolute stereochemistry of the enyne (26) controls stereochemistry of the product (27).[13]
Au and Pt mediated enyne cycloisomerization
Alkyne activation with π–acidic metals such as Au or Pt is a conventional method in complex organic manifold synthesis, however how this activation exacts reactivity is not fully understood and thus mechanism is largely proposed on the basis of reaction outcome and theoretical calculations.[14][15] Cationic Au(I) and Pt(II) catalyst are attractive choices as they display strong Lewis acid character and the ability to stabilize cationic intermediates while being bench stable.[16]
A versatile function of Au(I) catalyzed enyne cycloisomerization is the construction of asymmetric medium–sized rings, which is a challenge in the synthesis of ornamented molecular design. Convenient access to asymmetric 7– and 8–membered carbocycles is possible using chiral BINAP Au(I) gold catalyst, giving a wide variety of products.[17]
It is proposed that intramolecular cyclopropanation occurs via a 1,2–shift of the propargyl ester mediated by Au to give a syn–Au vinyl carbenoid species (29).[17] Computational studies show that the syn–intermediate, 29, is formed under kinetic control and it is suggested that it is in equilibrium with the thermodynamically favorable cis–intermediate which may be intercepted by a nucleophile leading to vinyl cyclopropane diene products, however this is beyond the scope of this article.[17]
Vinylcycloalkenes is another functional class of products accessible through alkyne activation of enynes with π–acidic metals. PtCl2 has been shown to catalyze the formation of a variety of exotic vinylcycloalkenes from readily accessible starting materials (figure 10).[18]
Notably, a ring expansion is observed for enynes with cyclic alkene motifs. This is rationalized by a formal insertion of the methylene group of the olefin between the two carbons of the alkyne; a mechanistic reasoning for this ring expansion has also been proposed.[19] The formation of these vinycloalkenes in concert with its ability to undergo a ring expansion was exploited to construct intermediate 36en route to streptorubin B.[18] A similar transformation is possible using cationic Au(I) complexes, however here one can select for vinycycloalkene products through a mechanism proceeding via an initial 5–exo–dig or bicyclopropanes can be produced via an initial 6–endo–dig.[20] It is suggested through DFT calculations that the 5–exo–dig cyclization is favored for Au(I) complexes as it has a lower activation barrier relative to the 6–endo–dig and indeed numerous examples of vinylcycloalkenes products produced via an initial 5–exo–dig are given (figure 11).
The reactivity can be reversed by careful selection of reaction conditions, catalyst selection and substrate.[20] The divergent reactivity of these transition metal catalyzed cycloisomerizations further demonstrates their synthetic utility in building unique molecular skeletons.
References
↑For a review on cycloisomerization reactions see: Enantioselective, transition metal catalyzed cycloisomerizations Angela Marinetti, Hélène Jullien and Arnaud Voituriez Chem. Soc. Rev., 2012,41, 4884-4908 doi:10.1039/C2CS35020C, Critical Review .
12345Watson, I. D. G.; Toste, F. D. Chem. Sci. 2012, 3, 2899–2919.
1234Nodes, W. J.; Nutt, D. R.; Chippindale, A. M.; Cobb, A. J. A. J. Am. Chem. Soc. 2009, 131, 16016–16017.
↑For a review on thiourea catalysis see: Zhang, Z.; Schreiner, P. R. Chem. Soc. Rev. 2009, 38, 1187–1198.
12Belmessieri, D.; Houpliere, A.; Calder, E. D. D.; Taylor, J. E.; Smith, A. D. Chem. Eur. J. 2014, 20, 9762–9769.
12Johnson, J. S.; Evan, D. A. J. Org. Chem. 1997, 62, 786–787.
↑Sizemore, N.; Rychnovsky, S. D. Org. Lett. 2014, 16, 688–691.
↑Izquierdo, J. Hutson, G. E.; Cohen, D. T.; Scheidt, K. Angew. Chem. Int. Ed. 2012, 51, 11686–11698.
↑For a review on N–heterocyclic carbene organocatalysis cascade reactions, see: Grossman, A.; Enders, D. Angew. Chem. Int. Ed. 2012, 51, 314–325.
123Kowalczyk, M.; Lupton, D. W. Angew. Chem. Int. Ed. 2014, 53, 5314–5317.
↑Genêt, J. –P.; Toullec, P. Y.; Michelet, V. Angew. Chem. Int. Ed. 2008, 47, 4268–4315.
12Newcomb, E. T.; Ferreira, E. M. Org. Lett. 2013, 15, 1772–1775.
↑Fürstner, A.; Davies, P. W. Angew. Chem. Int. Ed. 2007, 46, 3410–3449.
↑For a review on gold catalyzed enyne cycloisomerization reactions, see: Jiménez–Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350.
↑Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403.
123Watson, I. D. G.; Ritter, S; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2056–2057.
12Fürstner, A.; Stelzer, F.; Szillat, H. J. Am. Chem. Soc. 2001, 123, 11863–11869.
↑Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296.
12Nieto–Oberhuber, C.; Muñoz, M. P.; Buñel, E.; Nevado, C.; Cμrdenas, D. J.; Echavarren, A. M. Angew. Chem. Int. Ed. 2004, 43, 2402–2406.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.