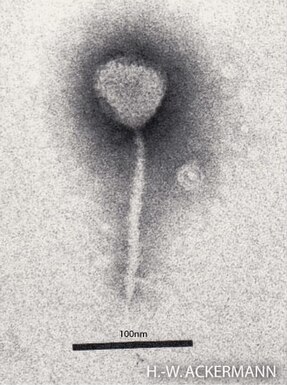
Enterobacteria phage λ is a bacterial virus, or bacteriophage, that infects the bacterial species Escherichia coli. It was discovered by Esther Lederberg in 1950. The wild type of this virus has a temperate life cycle that allows it to either reside within the genome of its host through lysogeny or enter into a lytic phase, during which it kills and lyses the cell to produce offspring. Lambda strains, mutated at specific sites, are unable to lysogenize cells; instead, they grow and enter the lytic cycle after superinfecting an already lysogenized cell.

In genetics, a promoter is a sequence of DNA to which proteins bind to initiate transcription of a single RNA transcript from the DNA downstream of the promoter. The RNA transcript may encode a protein (mRNA), or can have a function in and of itself, such as tRNA or rRNA. Promoters are located near the transcription start sites of genes, upstream on the DNA . Promoters can be about 100–1000 base pairs long, the sequence of which is highly dependent on the gene and product of transcription, type or class of RNA polymerase recruited to the site, and species of organism.

A transposable element is a DNA sequence that can change its position within a genome, sometimes creating or reversing mutations and altering the cell's genetic identity and genome size. Transposition often results in duplication of the same genetic material. Barbara McClintock's discovery of them earned her a Nobel Prize in 1983.

Transcription is the process of copying a segment of DNA into RNA. The segments of DNA transcribed into RNA molecules that can encode proteins are said to produce messenger RNA (mRNA). Other segments of DNA are copied into RNA molecules called non-coding RNAs (ncRNAs). Averaged over multiple cell types in a given tissue, the quantity of mRNA is more than 10 times the quantity of ncRNA. The general preponderance of mRNA in cells is valid even though less than 2% of the human genome can be transcribed into mRNA, while at least 80% of mammalian genomic DNA can be actively transcribed, with the majority of this 80% considered to be ncRNA.
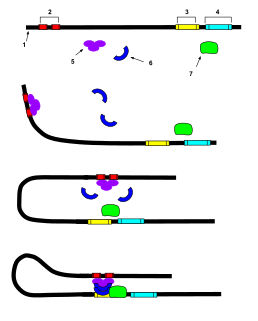
In genetics, an enhancer is a short region of DNA that can be bound by proteins (activators) to increase the likelihood that transcription of a particular gene will occur. These proteins are usually referred to as transcription factors. Enhancers are cis-acting. They can be located up to 1 Mbp away from the gene, upstream or downstream from the start site. There are hundreds of thousands of enhancers in the human genome. They are found in both prokaryotes and eukaryotes.
In molecular biology and genetics, transcriptional regulation is the means by which a cell regulates the conversion of DNA to RNA (transcription), thereby orchestrating gene activity. A single gene can be regulated in a range of ways, from altering the number of copies of RNA that are transcribed, to the temporal control of when the gene is transcribed. This control allows the cell or organism to respond to a variety of intra- and extracellular signals and thus mount a response. Some examples of this include producing the mRNA that encode enzymes to adapt to a change in a food source, producing the gene products involved in cell cycle specific activities, and producing the gene products responsible for cellular differentiation in multicellular eukaryotes, as studied in evolutionary developmental biology.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "gene-by-gene" approach.
The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.
In molecular biology, open reading frames (ORFs) are defined as spans of DNA sequence between the start and stop codons. Usually, this is considered within a studied region of a prokaryotic DNA sequence, where only one of the six possible reading frames will be 'open'. Such an ORF may contain a start codon and by definition cannot extend beyond a stop codon. That start codon indicates where translation may start. The transcription termination site is located after the ORF, beyond the translation stop codon. If transcription were to cease before the stop codon, an incomplete protein would be made during translation. In eukaryotic genes with multiple exons, introns are removed and exons are then joined together after transcription to yield the final mRNA for protein translation. In the context of gene finding, the start-stop definition of an ORF therefore only applies to spliced mRNAs, not genomic DNA, since introns may contain stop codons and/or cause shifts between reading frames. An alternative definition says that an ORF is a sequence that has a length divisible by three and is bounded by stop codons. This more general definition can be useful in the context of transcriptomics and metagenomics, where a start or stop codon may not be present in the obtained sequences. Such an ORF corresponds to parts of a gene rather than the complete gene.

The Encyclopedia of DNA Elements (ENCODE) is a public research project which aims to identify functional elements in the human genome.
Cis-regulatory elements (CREs) or Cis-regulatory modules (CRMs) are regions of non-coding DNA which regulate the transcription of neighboring genes. CREs are vital components of genetic regulatory networks, which in turn control morphogenesis, the development of anatomy, and other aspects of embryonic development, studied in evolutionary developmental biology.
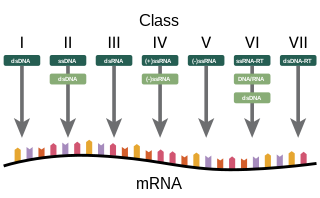
Baltimore classification is a system used to classify viruses based on their manner of messenger RNA (mRNA) synthesis. By organizing viruses based on their manner of mRNA production, it is possible to study viruses that behave similarly as a distinct group. Seven Baltimore groups are described that take into consideration whether the viral genome is made of deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), whether the genome is single- or double-stranded, and whether the sense of a single-stranded RNA genome is positive or negative.
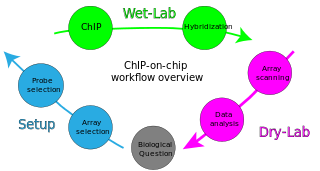
ChIP-on-chip is a technology that combines chromatin immunoprecipitation ('ChIP') with DNA microarray ("chip"). Like regular ChIP, ChIP-on-chip is used to investigate interactions between proteins and DNA in vivo. Specifically, it allows the identification of the cistrome, the sum of binding sites, for DNA-binding proteins on a genome-wide basis. Whole-genome analysis can be performed to determine the locations of binding sites for almost any protein of interest. As the name of the technique suggests, such proteins are generally those operating in the context of chromatin. The most prominent representatives of this class are transcription factors, replication-related proteins, like origin recognition complex protein (ORC), histones, their variants, and histone modifications.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.
Genomatix GmbH is a computational biology company headquartered in Munich, Germany, with a seat of business in Ann Arbor, Michigan, U.S.A.
Peak calling is a computational method used to identify areas in a genome that have been enriched with aligned reads as a consequence of performing a ChIP-sequencing or MeDIP-seq experiment. These areas are those where a protein interacts with DNA. When the protein is a transcription factor, the enriched area is its transcription factor binding site (TFBS). Popular software programs include MACS. Wilbanks and colleagues is a survey of the ChIP-seq peak callers, and Bailey et al. is a description of practical guidelines for peak calling in ChIP-seq data.
DNase-seq is a method in molecular biology used to identify the location of regulatory regions, based on the genome-wide sequencing of regions sensitive to cleavage by DNase I. FAIRE-Seq is a successor of DNase-seq for the genome-wide identification of accessible DNA regions in the genome. Both the protocols for identifying open chromatin regions have biases depending on underlying nucleosome structure. For example, FAIRE-seq provides higher tag counts at non-promoter regions. On the other hand, DNase-seq signal is higher at promoter regions, and DNase-seq has been shown to have better sensitivity than FAIRE-seq even at non-promoter regions.
TRANSFAC is a manually curated database of eukaryotic transcription factors, their genomic binding sites and DNA binding profiles. The contents of the database can be used to predict potential transcription factor binding sites.
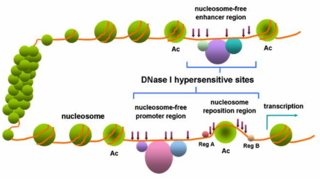
In genetics, DNase I hypersensitive sites (DHSs) are regions of chromatin that are sensitive to cleavage by the DNase I enzyme. In these specific regions of the genome, chromatin has lost its condensed structure, exposing the DNA and making it accessible. This raises the availability of DNA to degradation by enzymes, such as DNase I. These accessible chromatin zones are functionally related to transcriptional activity, since this remodeled state is necessary for the binding of proteins such as transcription factors.