Related Research Articles

A base pair (bp) is a fundamental unit of double-stranded nucleic acids consisting of two nucleobases bound to each other by hydrogen bonds. They form the building blocks of the DNA double helix and contribute to the folded structure of both DNA and RNA. Dictated by specific hydrogen bonding patterns, "Watson–Crick" base pairs allow the DNA helix to maintain a regular helical structure that is subtly dependent on its nucleotide sequence. The complementary nature of this based-paired structure provides a redundant copy of the genetic information encoded within each strand of DNA. The regular structure and data redundancy provided by the DNA double helix make DNA well suited to the storage of genetic information, while base-pairing between DNA and incoming nucleotides provides the mechanism through which DNA polymerase replicates DNA and RNA polymerase transcribes DNA into RNA. Many DNA-binding proteins can recognize specific base-pairing patterns that identify particular regulatory regions of genes.

Nucleotides are organic molecules consisting of a nucleoside and a phosphate. They serve as monomeric units of the nucleic acid polymers deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), both of which are essential biomolecules within all life-forms on Earth. Nucleotides are obtained in the diet and are also synthesized from common nutrients by the liver.

Polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation. It is fundamental to much of genetic testing including analysis of ancient samples of DNA and identification of infectious agents. Using PCR, copies of very small amounts of DNA sequences are exponentially amplified in a series of cycles of temperature changes. PCR is now a common and often indispensable technique used in medical laboratory research for a broad variety of applications including biomedical research and criminal forensics.

A primer is a short single-stranded nucleic acid utilized by all living organisms in the initiation of DNA synthesis. The enzymes responsible for DNA replication, DNA polymerases, are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable.
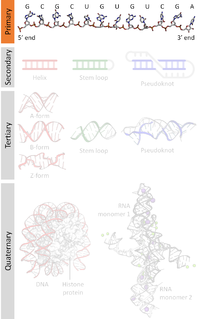
A nucleic acid sequence is a succession of bases signified by a series of a set of five different letters that indicate the order of nucleotides forming alleles within a DNA or RNA (GACU) molecule. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
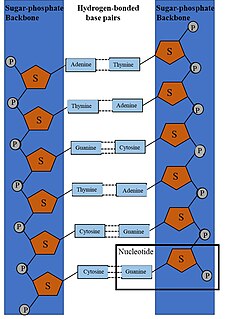
DNA synthesis is the natural or artificial creation of deoxyribonucleic acid (DNA) molecules. DNA is a macromolecule made up of nucleotide units, which are linked by covalent bonds and hydrogen bonds, in a repeating structure. DNA synthesis occurs when these nucelotide units are joined together to form DNA; this can occur artificially or naturally. Nucleotide units are made up of a nitrogenous base, pentose sugar (deoxyribose) and phosphate group. Each unit is joined when a covalent bond forms between its phosphate group and the pentose sugar of the next nucleotide, forming a sugar-phosphate backbone. DNA is a complementary, double stranded structure as specific base pairing occurs naturally when hydrogen bonds form between the nucleotide bases.

In molecular biology and genetics, GC-content is the percentage of nitrogenous bases in a DNA or RNA molecule that are either guanine (G) or cytosine (C). This measure indicates the proportion of G and C bases out of an implied four total bases, also including adenine and thymine in DNA and adenine and uracil in RNA.
A restriction digest is a procedure used in molecular biology to prepare DNA for analysis or other processing. It is sometimes termed DNA fragmentation. Hartl and Jones describe it this way:
This enzymatic technique can be used for cleaving DNA molecules at specific sites, ensuring that all DNA fragments that contain a particular sequence at a particular location have the same size; furthermore, each fragment that contains the desired sequence has the sequence located at exactly the same position within the fragment. The cleavage method makes use of an important class of DNA-cleaving enzymes isolated primarily from bacteria. These enzymes are called restriction endonucleases or restriction enzymes, and they are able to cleave DNA molecules at the positions at which particular short sequences of bases are present.
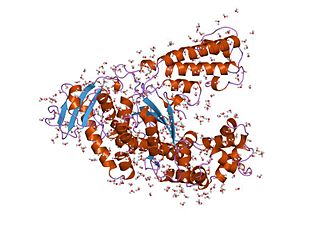
Taq polymerase is a thermostable DNA polymerase I named after the thermophilic eubacterial microorganism Thermus aquaticus, from which it was originally isolated by Chien et al. in 1976. Its name is often abbreviated to Taq or Taq pol. It is frequently used in the polymerase chain reaction (PCR), a method for greatly amplifying the quantity of short segments of DNA.
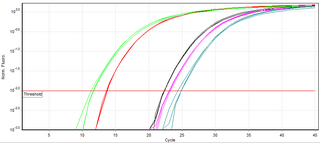
A real-time polymerase chain reaction, also known as quantitative Polymerase Chain Reaction (qPCR), is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.

Genetic analysis is the overall process of studying and researching in fields of science that involve genetics and molecular biology. There are a number of applications that are developed from this research, and these are also considered parts of the process. The base system of analysis revolves around general genetics. Basic studies include identification of genes and inherited disorders. This research has been conducted for centuries on both a large-scale physical observation basis and on a more microscopic scale. Genetic analysis can be used generally to describe methods both used in and resulting from the sciences of genetics and molecular biology, or to applications resulting from this research.
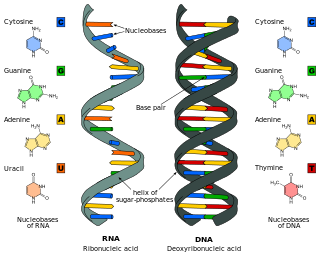
Nucleic acid analogues are compounds which are analogous to naturally occurring RNA and DNA, used in medicine and in molecular biology research. Nucleic acids are chains of nucleotides, which are composed of three parts: a phosphate backbone, a pentose sugar, either ribose or deoxyribose, and one of four nucleobases. An analogue may have any of these altered. Typically the analogue nucleobases confer, among other things, different base pairing and base stacking properties. Examples include universal bases, which can pair with all four canonical bases, and phosphate-sugar backbone analogues such as PNA, which affect the properties of the chain . Nucleic acid analogues are also called Xeno Nucleic Acid and represent one of the main pillars of xenobiology, the design of new-to-nature forms of life based on alternative biochemistries.
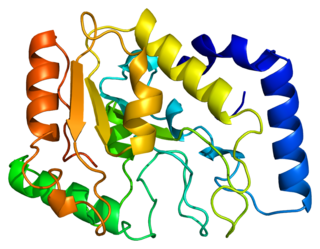
Uracil-DNA glycosylase, also known as UNG or UDG. Its most important function is to prevent mutagenesis by eliminating uracil from DNA molecules by cleaving the N-glycosidic bond and initiating the base-excision repair (BER) pathway.
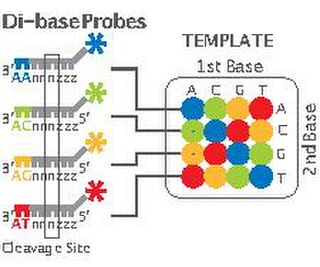
2 Base Encoding, also called SOLiD, is a next-generation sequencing technology developed by Applied Biosystems and has been commercially available since 2008. These technologies generate hundreds of thousands of small sequence reads at one time. Well-known examples of such DNA sequencing methods include 454 pyrosequencing, the Solexa system and the SOLiD system. These methods have reduced the cost from $0.01/base in 2004 to nearly $0.0001/base in 2006 and increased the sequencing capacity from 1,000,000 bases/machine/day in 2004 to more than 100,000,000 bases/machine/day in 2006.
The versatility of polymerase chain reaction (PCR) has led to a large number of variants of PCR.
The nucleic acid notation currently in use was first formalized by the International Union of Pure and Applied Chemistry (IUPAC) in 1970. This universally accepted notation uses the Roman characters G, C, A, and T, to represent the four nucleotides commonly found in deoxyribonucleic acids (DNA). Given the rapidly expanding role for genetic sequencing, synthesis, and analysis in biology, researchers have been compelled to develop alternate notations to further support the analysis and manipulation of genetic data. These notations generally exploit size, shape, and symmetry to accomplish these objectives.
TA cloning is a subcloning technique that avoids the use of restriction enzymes and is easier and quicker than traditional subcloning. The technique relies on the ability of adenine (A) and thymine (T) on different DNA fragments to hybridize and, in the presence of ligase, become ligated together. PCR products are usually amplified using Taq DNA polymerase which preferentially adds an adenine to the 3' end of the product. Such PCR amplified inserts are cloned into linearized vectors that have complementary 3' thymine overhangs.

Duplex sequencing is a library preparation and analysis method for next-generation sequencing (NGS) platforms that employ random tagging of double-stranded DNA to detect mutations with higher accuracy and lower error rate. This method uses degenerate molecular tags in addition to sequencing adapters to recognize reads originating from each strand of DNA. The generated sequencing reads then will be analyzed using two methods: single-strand consensus sequences (SSCSs) and Duplex consensus sequences (DCSs) assembly. Duplex sequencing theoretically can detect mutations with frequencies as low as 5 x 10−8 that is more than 10,000 fold higher in accuracy compared to the conventional next-generation sequencing methods.
Sequence saturation mutagenesis (SeSaM) is a chemo-enzymatic random mutagenesis method applied for the directed evolution of proteins and enzymes. It is one of the most common saturation mutagenesis techniques. In four PCR-based reaction steps, phosphorothioate nucleotides are inserted in the gene sequence, cleaved and the resulting fragments elongated by universal or degenerate nucleotides. These nucleotides are then replaced by standard nucleotides, allowing for a broad distribution of nucleic acid mutations spread over the gene sequence with a preference to transversions and with a unique focus on consecutive point mutations, both difficult to generate by other mutagenesis techniques. The technique was developed by Professor Ulrich Schwaneberg at Jacobs University Bremen and RWTH Aachen University.

Linear Amplification via Transposon Insertion (LIANTI) is a linear whole genome amplification (WGA) method. To analyze or sequence very small amount of DNA, i.e. genomic DNA from a single cell, the picograms of DNA is subject to WGA to amplify at least thousands of times into nanogram scale, before DNA analysis or sequencing can be carried out. Previous WGA methods use exponential/nonlinear amplification schemes, leading to bias accumulation and error propagation. LIANTI achieved linear amplification of the whole genome for the first time, enabling more uniform and accurate amplification.
References
- ↑ "Degenerate Bases & Spiking - Introduction". www.genelink.com. Retrieved 2017-10-07.
- ↑ "Can somebody explain to me what "spiking" means in RT-PCR and why do you - General Lab Techniques". www.protocol-online.org. Retrieved 2017-10-07.